Note: Descriptions are shown in the official language in which they were submitted.
81792342
FLOC COMPOSITION CONTAINING METAL IONS
This application is a divisional of CA 2,677,817, filed on February 14, 2008.
FIELD OF THE INVENTION
The Invention provides for large scale water purification that is useful
for removing anions and cations simultaneously from a large variety of
contaminated waters.
A particularly useful application is in the purification of acid mine
drainage.
BACKGROUND OF THE INVENTION
The demand for decontaminated fresh water has been steadily
increasing in the United States and world wide due to increasing populations
and increasing industrialization, mining operations, and agriculture and this
trend is projected to continue. In addition, fresh water sources, such as
wells
that use aquifer water are increasingly found to be contaminated by these
mostly human activities. A particularly problematic example is that of mining,
especially that of coal mining, where the mining activity has exposed gangue
minerals left in the mine and mine tailings to erosion by air, water, and
microbial action. Sulfldic minerals, such as pyrite, FeS2, are commonly found
in many geological strata, and especially in reducing ore bodies such as coals
and metal sulfide.
As described thoroughly in an extensive literature spanning many
decades, coal, metal and other mining operations, and natural weathering
fissures -have allowed water, air, and microbial arress to these reducing
substances. These conditions promote the oxidation of the sulfidic minerals to
1
CA 2911119 2018-03-05
CA 02911119 2015-11-03
' 27728-2D1
water soluble ferrous sulfate and other metal sulfates; thereby, especially
when exposed to air at the surface, producing an acidic discharge or ground
water of hundreds and even often above 2000 ppm of total dissolved solids
(see Tables 2A, 2B, and 2C). Such contaminated ground water is unsuitable
s for most uses, including municipal, industrial, residential, and farming;
is foul
tasting; is odorous; is toxic to aquatic ecosystems, plants, and animals; and
can mobilize additional metal contaminants by acid dissolution of natural or
man-made materials.
The problems of water purification and treatments of acid mine
io drainage and acid rock drainage, collectively referred to as acid mine
drainage
or AMD water, are well described in the literature. Hereinafter, when acid
mine drainage is discussed, unless mentioned otherwise, the text will apply to
acid rock and natural gas well brines drainage also. Conventional
technologies have been found to be unsuitable for processing such waters
15 especially where total flow rates exceed 10 gal/min and can reach
100,000
gal/min. Dissolved metal cation and counter anion concentrations in acid
mine drainage can be far above the levels removable by technologies known
in the art. Other conventional technologies have major disadvantages
including high initial capital costs, slow reaction times, high reagent costs,
20 reagent hazards, and/or production of waste sludges. But most
importantly,
they do not remove the major problematic contaminant, sulfate ion. Sulfate
ion is responsible for heavy fouling or scaling during industrial use, foul
tastes
and odors, laxative effects, and very high level of corrosiveness to
construction metals and concrete.
BRIEF DESCRIPTION OF THE INVENTION
Some embodiments of the invention may provide for a practical, low-cost,
large-scale water purification process with minimal or no waste generation,
suitable for very high flow rates, even thousands of gallons per minute,
provide
simultaneous, very rapid anion and cation removal,including sulfate ion, which
thereby reduces total dissolved solids (TDS), and features a small equipment
foot print,
and co-product production. Some embodiments of the invention are particularly
suitable
2
CA 02911119 2015-11-03
27728-2D1
for the purification of acid mine drainage or acid rock ground drainage waters
(AMD/ARD) without the massive waste generation characteristic of prior art
technologies. The new process of some embodiments of the invention provides
recovery
of non-toxic, low TDS, and useful purified water with metals and sulfate co-
products.
Some embodiments of the invention provide a means to substantially purify
water,
especially suited for large volumes of water in a continuous fashion, in a
unique manner
by the simultaneous removal of anions and cations using a process based on
a particular combination of liquid-liquid extraction (LLX), oil skimming, and
flotation (F) technologies. Unique attributes of the process chemistry and
io associated device design of some embodiments of the invention, referred
to as flotation
liquid-liquid extraction (F-LLX) (typically the floc and extractant float),
are that it:
= Provides for the fast removal of these ions at rates 10-100 times faster
than conventional technologies,
= Where only 45 to 90 seconds of contact time is required, and
= Enables the extraction of metal ions normally not extractable or not well
extracted by conventional methods,
= Extracts a broader spectrum of ions, and
= Extracts anions and cations simultaneously (TDS) to lower residual
concentrations than by conventional LLX processing.
= Performs these extractions at mild conditions of pH and temperature
(nominally pH 7-8 and ambient temperature of 1 C to 99 C).
= Provides the production of metal ion product concentrates, solid salts,
and/or solids of oxides, hydroxides and/or carbonates.
= Provides the production of sulfate ion product concentrates and/or solid
salts.
= Provides a low cost means to purify large volumes of water continuously
and in high yield (>99%), even thousands of gallons per minute in a
simple manner, and with relatively much smaller equipment size than
conventional treatments.
= Provides a low cost means to purify continuously large volumes of water
that are contaminated with sulfate ion (300-10,000 mg/L 5042-), and even
thousands of gallons (0.5-10 Kgal/min) per minute.
3
CA 02911119 2015-11-03
27728-2D1
= Provides a one step, high yielding, and continuous unit operation that
removes toxic cations, salinity, hardness, TSS, TDS, and/or acidity,
including the very difficult species: ferric, ferrous, aluminum, nickel,
cobalt, manganese, zinc and sulfate ions with acid neutralization, in a
relatively small size of hardware with concomitant concentration of values
to enable their use as products.
For a typical acid mine drainage stream, the process of some embodiments of
the
invention does not produce large volumes of gypsum/limestone sludge wastes,
nor does it
produce membrane or resin back flush wastes, which are produced by
conventional technologies. Some embodiments of the invention accomplish waste
prevention
by avoiding the addition of voluminous amounts of limestone, lime, slaked
lime,
dolomite, etc., minerals or wetland muds and vegetation. Instead, an
important attribute of some embodiments of the invention is that it handles
the waters for only
a few minutes and then releases it; while at the same time some embodiments of
the invention
simultaneously concentrates the contaminants many fold, normally 10 to 1000
or more times, while continuously recycling the water-immiscible liquid
extractant phase. Note that the term extractant phase and extractant
solution are used interchangeably herein. The continuous regeneration of the
extractant phase results in highly-efficient use of working capital thereby
zo requiring only a small inventory of the extractant phase. The metal and
sulfate product concentrates are readily processed into useful commodities by
well-known methods.
Some embodiments of the invention include new compositions of matter
consisting of
formulations of at least one cationic liquid extractant component combined
with metal ions to form a colloid consisting of one or more ions selected from
oxide, hydroxide carbonate and/or bicarbonate ions combined with one or
more non-basic anions to form an oil soluble phase that, when combined,
form a floc useful for the simultaneous separation and recovery of such
solutes. It is especially useful to reduce high metal ion and sulfate ion
concentrations in water, for example as are found in mining and mineral
processing effluent waters, in industrial process effluent waters, livestock
farm waters, and the like. TDS is reduced from high levels, for example TDS
4
CA 02911119 2015-11-03
' 27728-2D1
values of 700-5000 mg/L, down to levels that allow water re-use, for example
to less than 600 mg/L TDS for surface water discharge, or less than 250 ppm
total dissolved solids to enable re-use as drinking/potable water, and down to
deionized water for industrial or non H2S-forming potable water use where
TDS less than about 125 ppm is needed, and with toxic metal ions removed
to < 1 or even <0.3 ppm. Some embodiments of the invention can reduce sulfate
ion
to <250 or even to <30 PPm=
It is believed that the above-mentioned efficient deionization of water
is accomplished by a unique physio-chemical process based on involving the
lo fast formation of a new substantially hydrophobic floc material
described
above and where such material can be in colloidal form, particulate form, gel
form, and/or floc form, and preferably also contain oil soluble non-flammable
alcohol, ester and/or other oxygenated hydrocarbon modifier, with a low-
viscosity, non-flammable, hydrocarbon liquid diluent, and that is less dense
than water and is immiscible in water so that, left unmixed, it readily
separates from water by spontaneous phase disengagement.
Production of waste is minimized in some embodiments of the invention by
avoiding
the use of conventional bulking reagents in current practice such as lime,
slaked lime,
limestone, dolomite, or ferric, ferrous, salts, and/or aluminum salts or
coagulants and/or precipitants, that invariably result in producing hazardous,
unuseable, low-density solid sludge toxic wastes that lead to dilution of the
values.
New products of commerce are also provided that are new composition
of matter of the formulas:
(for the case of divalent metal ions, N) ....(a)
and
(for the case of trivalent metal ions, M) ....(b)
and consists of a combination of ferric, ferrous, aluminum, and sulfate ions
in
specific range of proportions. This material is formed using some embodiments
of the
invention as highly concentrated aqueous solutions ready for use and/or as a
crystalline
5
CA 02911119 2015-11-03
27728-2D1
solid material. This new composition is useful for purification of potable
water; purification of agricultural liquid wastes, food wastes, and municipal
sewage; purification of industrial waste water, and the like.
The following disclosure contains a general description where steps of
one or more processes are designated as first, second, third, and so on. This
is solely to clearly differentiate process steps that may be repeated in
different embodiments from each other.
Some embodiments of the invention include a method for purifying aqueous
io solutions to remove ionic components, consisting of at least one cation
and
one anion, comprising:
a. mixing the aqueous solution with an extractant phase for
typically up to 30 min, preferably 10 min, more preferably 3 min, and
most preferably 45-90 seconds to form an unstable emulsion wherein
the extractant phase comprises;
(i). A cationic extractant that contains a basic moiety that forms
a neutral to anionic floc with at least one or more of the cationic
components of the aqueous solution and with at least one
anionic component of the aqueous solution, wherein the
extractant comprises a positively charged moiety having at least
8 carbon atoms, preferably 18 carbon atoms, and most
preferably 25 carbon atoms, up to about 34 carbon atoms,
wherein the carbon atoms are present as hydrocarbons, and a
moiety comprising an anionic base;
(iii). an optional nonflammable hydrocarbon diluent;
(iv). an optional nonflammable oxygenated hydrocarbon
modifier for enhancing phase disengagement rate and/or
minimizing water content of the cation and anion loaded
extractant portion of the unstable emulsion;
(v) wherein the equilibrium pH of the unstable emulsion is about
2 to about 12 and is controlled by the initial phase ratio of the
extractant and aqueous phases;
6
CA 02911119 2015-11-03
=
27728-2D1
b. disengaging a first treated aqueous phase and a first loaded
extractant phase from the unstable emulsion, wherein a colloid and/or
floc and/or water immiscible precipitate forms in the first loaded
extractant phase; and
c. separating the first loaded extractant phase and floc from the
first treated aqueous phase, wherein the first treated aqueous phase
comprises first purified water.
Typically the positively charged extractant component comprises a
quaternary ammonium and/or phosphonium compound selected from the
io group consisting of R4N+, FLIP+, and/or an alkylated monoguanadinium
compound; where the R groups may differ and are a hydrocarbon consisting
of alkyl groups, aryl groups, alkylaryl groups, any combination of these,
including atoms of other elements such as N, P, 0 and S so that the water
solubility is not significantly increased or the monocationic charge for the
is whole molecule is not changed, and the charge does not change with pH
up
until a pH of about 10, and preferably a pH of about 12, and where the
minimum carbon number (CN) is >8, preferably > 17, and more preferably
> 24 up to a total of about 34, and most preferably where at least one alkyl
group in the molecule is branched and/or the whole molecule has a tripodal
20 structure.
Typically the anionic base is selected from the group consisting of
C032- , HCO3-, or OH-, but, depending on the desired products produced and
water contaminants present, also could include P043-, HP042-, or H2PO4- ,
or S2-
25 Some embodiments of the invention provide the unique ability to
reduce the total
dissolved solid levels of waters by simultaneously co-extracting at least one
each of the
anionic and cationic components present in the water, and are captured from
the aqueous solution in the first loaded extractant phase and floc and the
anionic components are one or more of the group consisting of sulfate,
30 selenate, nitrate, nitrite, phosphate, arsenate, arsenite, bromate,
bromide,
perchlorate, iodide, chloride, chromate(VI), permanganate, bisulfide, and
sulfide ions, including the protonated versions of these ions, including those
7
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
protonated species that would render the ion neutral, and any combination
and concentrating level of these ions.
Typically the removed cationic component is a metal ion, most often
selected from one or more of the group consisting of Ni+2, Fen cu2+,
Ag+, Zn2+, Co2+, Com, Fe2+, ca2+, mg2+, 0:12+, mn2+, pb2+, Hg2+, Hg22+,
CH3Hg+, and CrlII, wherein Roman numerals represent variable speciation and
the others represent normally aqueous ions.
A further embodiment includes neutralization of the acidic component
of the water simultaneously with the co-extraction of metal ions and anions
lci into the extractant phase (E-phase).
A further embodiment includes stripping metal ions and co-extracted
anions from the separated first loaded extractant phase, preferably as it
exits
the extraction decanter, by the steps of:
a. mixing the separated first loaded extractant phase, colloids, and
floc with an aqueous acid to form a second unstable emulsion; wherein metal
ions in the separated first loaded extractant phase and floc are stripped from
the emulsion colloids and/or floc of loaded extractant phase making up the
unstable emulsion and dissolved into the aqueous acid phase, and
b. disengaging a first loaded aqueous acid phase and a metal ion
stripped extractant phase from the second unstable emulsion and where the
first stripped extractant phase contains at least a portion of the anions
extracted into the first loaded extractant phase.
A yet further embodiment includes mixing a metal ion stripped
.. extractant phase with an aqueous solution of anionic base selected from the
group consisting of C032- , HCO3- , OH- , P043-, HP042-or H21304- , H5, or S2-
,
wherein HCO3, C032- and OH- are most preferred, and where the bicarbonate,
carbonate, and/or hydroxide ion loaded extractant phase produces a third
unstable emulsion, wherein anion, sulfate ion in the case of AMD feed water,
is stripped from the metal ion stripped extractant phase into an aqueous
phase and producing a bicarbonate, carbonate and/or hydroxide loaded
extractant solution; disengaging the loaded bicarbonate, carbonate and/or
8
CA 02911119 2015-11-03
WO 20081100610
PCT/US2008/002092
hydroxide loaded extractant phase to yield a regenerated extractant phase
stripped of at least a portion of the sulfate content, and normally stripped
of
more than 90% of its sulfate ion content and preferably stripped of more than
98% of its sulfate ion content, and a third aqueous phase of alkali metal ion,
anion solution, alkali, sulfate in the case of AMD water, and/or a slurry of
alkali metal ion sulfate and/or alkaline earth metal ion sulfate solution, for
the
case of alkaline earth Mg, and slurry in the case of alkaline earth Ca, or a
combination of these; and separating the loaded carbonate, bicarbonate
and/or hydroxide regenerated extractant phase from the aqueous phase
containing the sulfate ion and any remaining carbonate or hydroxide ions,
where preferably most of the loaded basic ion is carbonate where the
extraction pH is less than 9 and at least a portion as hydroxide ion where the
extraction pH is >9, and where the bicarbonate ion component is <10% of
the basic anion component.
A yet further embodiment includes a method for treating an aqueous
solution to remove one or more ionic components comprising:
a. mixing at least a portion of the first treated aqueous phase
from the method above with an extractant phase to form a fourth
unstable emulsion wherein the extractant phase comprises;
(i). an extractant that forms a floc with one or more of the ionic
components of the aqueous solution, wherein the extractant
comprises a positively charged moiety having at least 8 carbon
atoms, and a component comprising an anionic base;
(iii). an optional medium to low viscosity, nonflammable
diluent;
(iv). an optional modifier for increasing phase disengagement
rate and completeness; wherein the equilibrium pH of the
unstable emulsion is about 5 to about 9.
b. disengaging a fourth treated aqueous phase and a second
loaded extractant phase from the fourth unstable emulsion, wherein a
floc forms in the second loaded extractant phase; and
9
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
c. separating the second loaded extractant phase and floc from
the second extracted aqueous phase, wherein the second extracted
aqueous phase comprises second purified water.
d. the above steps are repeated twice more to produce a
purified water that has been extracted four times.
Typically this method provides for optional steps where the fourth
purified water is further purified in one or more of an oil/water separator, a
solid/liquid separator, and/or organic odor sorbent to obtain a further fourth
purified water lower in at least one ion from the ions still remaining in the
first
io purified water, a purified water that is more neutral to slightly
alkaline pH,
and preferably lower in more than one ion selected from the list Ni+2, Cu+2,
Fe", = .+3,
Zn2+, CO2, COI", Fe2+, ca24., mg2+, mn2+, pb2+, Hg2+, Hg22+,
CH3Hg+, Crm, and the like, wherein Roman numerals represent variable
speciation and the others represent normally aqueous ions.
The method also provides for removing ionic species wherein the
removed ionic component is a metal ion selected from one or more of the
group consisting of cations Ni+2, Fein, A1+3, zn2+, co2+, Co", Fe2+, ca2+,
mg2+,
cd2+, mn2+, pb2+, Hg2+, Hg_22+, CH3Hg+, cu2+, cu2+, Cr", Au (I and III) and
the like, wherein Roman numerals represent variable speciation and the
others represent normally aqueous ions, and anions including one or more of
sulfate, selenate, nitrate, nitrite, phosphate, arsenate, arsenite, bromate,
bromide, perchlorate, iodide, chloride, chromate(VI), permanganate,
molybdate, vanadate, and sulfide ions, including the protonated versions of
these ions, including those protonated species that would render the ion
neutral, and any combination of these ions.
Typically the method provides for capturing at least one ionic
component is captured in the second loaded extractant phase and floc
(consisting of colloids, floc, and/or oil dispersible precipitate) and one or
more
of the group consisting of cations Ni+2, Fe", Al+3, zn2+, 032+, Co", Fe2+,
Mg2+, Cd2+, mn2+, pb2+, Hg2+, H 22+,
g CH3Hg+, Ag+, Au(I or III), Cu, Cu,
and/or Cr", wherein Roman numerals represent variable speciation and the
others represent normally aqueous ions, and anions including one or more of
CA 02911119 2015-11-03
' WO 20081100610
PCT/US2008/002092
sulfate, selenate, nitrate, nitrite, phosphate, organophosphonate, arsenate,
arsenite, bromate, bromide, perchlorate, iodide, chloride, chromate(VI),
permanganate, molybdate, vanadate, and sulfide ions, including the
protonated versions of these ions, for those ions not forming strong mineral
acids, including those protonated species that would render the ion neutral,
and any combination of these ions.
Other embodiments provide for a method wherein the first and/or
second loaded extractant phase and floc are further treated to separate
residual water from the loaded extractant phase and floc using a decanter
fitted with internal weirs to guide the surface flow of extractant phase with
floc in a narrowing channel such that the flow of the floc is maintained as it
thickens, and where the flow is maintained until it reaches and flows over an
overflow weir designed to promote such flow of thickened flocs, most
preferably by a about a 15 to about a 45 angle, from the vertical, approach
ramp, preferably smooth lip and about a 15 to about 45 exit ramp.
Another embodiment provides for stripping metal ions from at least a
portion of the separated second loaded extractant phase by the steps of:
a. mixing the separated second loaded extractant phase and floc with
an aqueous acid to form a fifth unstable emulsion; wherein metal ions in the
separated second loaded extractant phase and floc are stripped into the
aqueous acid phase, and
b. disengaging a fifth unstable emulsion with a second aqueous acid
phase to form a second metal ion stripped extractant phase separated from
the fifth unstable emulsion.
c. optionally recycling the second loaded aqueous strip acid phase to
contact additional volumes of the second loaded extractant phase.
Another embodiment provides for treating the second loaded aqueous
acid phase by one or more of an oil/water separator, a solid/liquid separator,
and/or an organic odor sorbent wherein a metal ion salt product is obtained.
11
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
Another typical method provides for purifying an aqueous solution to
remove one or more ionic components, especially residual manganese ion as
MnCO3 particulate, comprising:
a. mixing at least a portion of the second treated aqueous phase
from the method above with a basic extractant phase to form a sixth
unstable emulsion wherein the extractant phase comprises;
(i). an extractant that forms a floc with one or more of the ionic
components of the aqueous solution, wherein the extractant
comprises a positively charged moiety having at least 8 carbon
atoms, and a moiety comprising an anionic base;
(iii). an optional diluent;
(iv). an optional modifier for modifying phase disengagement;
wherein the equilibrium pH of the unstable emulsion is about
8.5 to about 10.5.
b. disengaging a third (or more) purified aqueous phase and a
third loaded extractant phase from the seventh unstable emulsion,
wherein a solid suspension and/or floc forms in the third loaded
extractant phase; and
c. separating the third loaded extractant phase and floc from the
third purified aqueous phase, wherein the third treated aqueous phase
comprises a third purified water.
A yet further method includes purifying an aqueous solution at higher
pH to remove one or more ionic components, especially magnesium ion as
Mg(OH)2 particulate, and any residual amount of sulfate ion, comprising:
a. mixing at least a portion of the third treated aqueous phase
from the above with a base-loaded extractant phase to form an eighth
unstable emulsion wherein the extractant phase comprises;
(i). an extractant that forms a floc or particulate with one or
more of the ionic components of the aqueous solution, wherein
the extractant comprises a positively charged moiety having at
least 8 carbon atoms, and a moiety comprising an anionic base,
most preferably hydroxide ion;
12
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
(iii). an optional diluent;
(iv). an optional modifier for modifying phase disengagement;
wherein the equilibrium pH of the unstable emulsion is about
10.5 to about 12.
b. disengaging an fourth treated aqueous phase and a fourth
loaded extractant phase from the eighth unstable emulsion, wherein a
floc and/or particulate slurry forms in the fourth loaded extractant
phase; and
c. separating the fourth loaded extractant phase and floc
and/or particulate from the fourth purified aqueous phase, wherein the
fourth purified aqueous phase comprises the eighth purified water.
Typically the method provides for treatment wherein the fourth purified
water is further treated in one or more of an oil/water separator, a
solid/liquid separator, to separate out the Mg(OH) 3 particulate product, to
obtain a further fourth purified water low in most ions, including cations
Ni+2,
Fern' Al+3, zn2+, 0)2+, Co", Fe2+, ca2+, mg2+, cd2+, mn2+, pb2+, Hg2+, cH3Hg+,
and/or CrIIL, wherein Roman numerals represent variable speciation and the
others represent normally aqueous ions; and anions including one or more of
sulfate, selenate, nitrate, nitrite, phosphate, arsenate, arsenite, bromate,
bromide, perchlorate, iodide, chloride, chromate(VI), permanganate, and
sulfide ions, including the protonated versions of these ions, including those
protonated species that would render the ion neutral, and any combination of
these ions, and a ninth separated extractant phase that is optionally sent to
the mixing step of a previous step to load even higher concentrations of
cation and anion values.
Another embodiment of the invention provides a method for treating
an aqueous solution containing ionic components comprising:
a. mixing the aqueous solution with a first extractant phase to
form a first unstable emulsion wherein the first extractant phase
comprises;
13
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
,
(i). one or more of a quaternary ammonium compound
comprising R4N+, an alkylated guanidinium compound, or a
quaternary phosphonium compound;
(ii) a carbonate and/or hydroxide component;
(iii). an optional diluent; and
(iv). an optional modifier for helping the phases disengage;
wherein the equilibrium pH of the unstable emulsion is about 2
to about 12.
b. disengaging a first treated aqueous phase and a first loaded
extractant phase from the unstable emulsion, wherein a floc forms in
the first loaded extractant phase; and
c. separating the first loaded extractant phase and floc from the
first treated aqueous phase, wherein the first treated aqueous phase is
purified water.
A further embodiment provides for a floc composition comprising:
an oil soluble cation, a metal ion, a hydroxide and or oxide, and an anion
according to the formula,
{(R4N+}x(N(IiDn(OH-):(5042-}w } m (for the case of divalent metal ions)
Or
{(R4Nix(M(III)MOHI(S042-)w } q (for the case of trivalent metal ions), and
mixtures of compositions of the formuli;
wherein x= 2-4, n = 0-1, Y = 1-2, Z= 2, and w = 1-3, and wherein M can be
Fe(III) or Al, and N can be Fe(II), Ni, Co(II), Cu, Zn, Pb, Cd, or Mn;
and wherein m and q can be 1 to about 100,000.
An additional floc composition includes a floc composition comprising:
an oil soluble cation, a metal ion, a hydroxide and or oxide, and an anion of
the formula,
(R4N1x(N(IIDn(Ohl-MS042-)w (for the case of divalent metal ions N)
or
(for the case of trivalent metal ions)
14
CA 02911119 2015-11-03
27728-2D1
and mixtures of materials according to the two formuli;
wherein x= 2-4, n = 0-1, Y = 1-2, Z= 2, and w = 1-3, and wherein M can be
Fe(III) or Al, and N can be Fe(II), Ni, Co(II), Cu, Zn, Pb, Cd, or Mn, and
where the material is dispersed in a non water soluble liquid such that the
floc
represents 1 to 100% by weight of the slurry.
A yet further embodiment includes a mixture of metal sulfates of the
the composition Fein, Fey Alz(SO4), where x=0.1, y=0.8 and z=0.1, giving a
value for w = (0.3+1.6+0.3)/2 = 1.1, or Feill03Feli1.6A10.004)1.1 for a
formula weight of 159 g/mole; and having the range of ratios: Fem0.03Fell.95
A10.03(SO4)1.04 (FW of 155 g/mole), to Fe1110.95Fen0.03A10.5(SO4)2.2(FW of 280
g/mole). Typically the material, Felll, Fey Alz(SO4), , produced by the
process of the invention from acid mine drainage feed water is in an aqueous
solution and has a dry weight of at least 0.1%, and preferably 1 to 5 %, and
most preferably 5 to 20%.
An additional embodiment of the invention includes an apparatus for
separating floc consisting of a. a decanter or extractor compartment having
an inlet for a mixture at one end of the compartment and an outlet for
aqueous spaced apart from the inlet; and b. a floc weir spaced apart from
the inlet, the floc weir comprising a tapered entrance ramp, a rounded lip
adjacent to the tapered entrance ramp and a floc weir exit slide adjacent to
and spaced beyond the rounded lip, wherein floc flows out of the
compartment over the entrance ramp, rounded lip and floc weir exit slide.
Typically the entrance ramp is tapered at 15 to 45 degrees from the vertical,
and the exit slide is tapered at 15 to 45 degrees from the vertical. A curved
drip point at the lowest point of the exit slide is typically used to provide
for
smooth separation of the floc from the slide. A rounded lip located between
the entrance ramp and exit slide of the floc weir Provides a smooth transition
for floc flow over this portion of the floc weir, and is typically the highest
point
on the floc weir.
81792342
According to another embodiment of the invention, there is provided a floc
composition comprising: an oil soluble cation, a metal ion, a hydroxide or
oxide, and an
anion of the formula, {(R4N+)),(N(11))n(OH)z(S042-)w}m for the case of
divalent metal ions or
{(R4N)0(111)M0FIVS042),,}q for the case of trivalent metal ions, and mixtures
thereof;
wherein x=2-4, n=0-1, Y=1-2, Z=2, and w=1-3, and wherein M is Fe(III) or Al,
and N is
Fe(II), Ni, Co(II), Cu, Zn, Pb, Cd, or Mn; and wherein m and q are each 1 to
about
100,000.
According to another embodiment of the invention, there is provided a floc
composition comprising: an oil soluble cation, a metal ion, a hydroxide or
oxide, and an
anion of the formula, (R4N+)x(N(11))401-1)0042-),, for the case of divalent
metal ions N or
(R4N+)õ(M(111))y(01-1),(8042-)w for the case of trivalent metal ions, and
mixtures thereof;
wherein x=2-4, n=0-1, Y=1-2, Z=2, and w=1-3, and wherein M is Fe(III) or Al,
and N is
Fe(ll), Ni, Co(II), Cu, Zn, Pb, Cd, or Mn, and where the material is dispersed
in a non-
water soluble liquid such that the floc represents 1 to 100% by weight of the
slurry.
15a
CA 2911119 2018-03-05
CA 02911119 2015-11-03
' 27728-2D1
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1A is a schematic drawing illustrating one part of the overall
flow materials of one embodiment of the invention.
Figure 18 is a schematic drawing illustrating a second part of the
s overall flow of materials of one embodiment of the invention. Figure 1A
and Figure 1B
taken together are intended to form one large figure.
Figure 1C is a schematic showing the general process steps involved in
a broad embodiment of the invention.
Figure 2 is a schematic illustrating the process for one embodiment of
the invention for the separation of trivalent and divalent metal ions and
sulfate ions, simultaneously from contaminated water. Also see the other
figures and examples illustrating more detailed aspects of the process.
Figure 3 is schematic diagram showing one typical LLX process flow
diagram of one embodiment of the invention illustrating a most preferred
apparatus
and process for treatment of acid mine drainage. Figure 3 also illustrates the
configuration used in Example 5 (Run #7).
Figure 4 is a schematic illustrating various aspects of a typical flotation
weir device according to one embodiment of the invention.
Figure 5 is a schematic of a flow guide that guides and facilitates the
flow of floc and extractant over a weir including details of its use in
one embodiment of the invention.
Figure 5A is a schematic drawing showing a side cutaway view of
Settler 526 with a plate for a flow guide.
Figure 58 is a schematic drawing showing a side cutaway of view of
Settler 526 with a block for a flow guide.
Figure 5C is a schematic drawing of a mixer according to the
embodiment of Figure 18.
Figure 6 is a schematic illustrating a typical underflow weir for
extractant introduction. =
Figure 7 is a schematic illustrating a typical design having extractant
solution introduction by tubing that is inside the mixing chamber.
16
CA 02911119 2015-11-03
27728-2W
4
Figure 8 is a schematic drawing illustrating a typical design for an
extractant storage chamber and the path of introducing extractant into the
mixing.
Figure 9 is a graph showing sulfate loading stages determination from
McCabe-Thiele plots with 5 (v/v)% extractant.
Figure 10 is similar to Figure 9 except that sulfate loading stages
determinations from McCabe-Thiele plots with 5 (v/v)% extractant are shown.
Figure 11 is a graph showing other conditions for sulfate loading stages
determination from McCabe-Thiele plots with 5 (v/v)% extractant.
io Figure 12 is similar to Figure 11 except that sulfate loading stages
determinations from McCabe-Thiele plots with 5 (v/v)% extractant are shown.
Figure 13 is a schematic diagram illustrating a sulfate circuit process
flow scheme according to one embodiment of the invention.
Figure 14 is a schematic diagram illustrating acid mine drainage
process flow scheme 1.
Figure 15 is a schematic diagram illustrating an acid mine drainage
process flow scheme 2.
Figure 16 is a plot of the correlation of AliquatTM and isodecanol
concentration and the impact of the formulation to post phase separation
zo water entrainment.
Figure 17 is a schematic drawing illustrating a presently preferred
configuration.
DETAILED DESCRIPTION OF THE INVENTION BEST MODE
Broadly, one embodiment of the invention provides for a method for purifying
an aqueous solution by simultaneously removing both cationic and anionic
components -
while neutralizing acidity and lowering total dissolved solids by the steps of
mixing the aqueous solution with a water immiscible liquid extractant phase
(E-phase) containing a basic moiety to form an unstable, high surface area
emulsion during which time these components transfer from the aqueous
phase to the E-phase, and acidity in the aqueous phase in neutralized by
transfer of at least a portion of the basic moiety to the aqueous phase. The
17
CA 02911119 2015-11-03
= WO 2008/100610
PCT/US2008/002092
extractant phase is made up of one or more of an cationic extractant
compound, most preferably one or more of a oil soluble quaternary
ammonium compound, comprising RIN1+, but also could be an alkylated
guanidinium compound, R5CN3H+,or an alkylated quaternary phosphonium
compound, R41)+, or a blend of these cationic compounds; wherein the
associated anion is a pH basic component. Typically the equilibrium pH of the
unstable emulsion is controllable from about 2 to about 13; the equilibrium
pH of the unstable emulsion is typically about 3 to about 9, or from 4 to
about
11 depending on desired ionic components for removal. This pH control is
io achieved by varying the E/A ratio, with pH increasing with this ratio.
Also, the
extractant alkyl groups can also be aryl, and/or alkylaryl.
If desired, more preferably, one or more of an optional hydrocarbon
diluent and/or a modifier, most preferably nonflammable, for promoting the
aqueous and extractant phases to remain fluid, disengage and dewater in a
following settling step, and the extractant phase homogenate, may be added
as part of the extractant phase; disengaging mixture of a treated aqueous
phase and a component-loaded extractant phase from the unstable emulsion,
wherein a new stable emulsion of floc phase forms in the loaded extractant of
a new composition of matter phase, and wherein the extractant phase and
aqueous phase are at least partially separated by allowing gravity separation,
optionally sped up by centrifugation and/or use with a hydrocyclone; and
where the floc remains in the E-phase but may tend to settle to the interface
with the aqueous phase, and separating the loaded extractant phase and floc
from the aqueous phase by using a skimming weir design to ensure that the
floc is removed from the aqueous phase along with the E-phase and before it
thickens into a slow to non-flowing solid precipitate.
The R groups in the quaternary ammonium compound, the
guanadinium compound, or the quaternary phosphonium compound can be
different hydrocarbons that have a carbon atom content of at least one each
and with a total carbon number for the R.41V+ compound of and/or R413+
compound 37 carbon atoms with a minimum total carbon number of nine, but
preferably 25. The basic requirement is that the "R" alkyl, alkylaryl and/aryl
18
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
groups provide sufficient hydrophobic character to the extractant phase so
that it remains a separate phase from the aqueous phase whether loaded
with floc or basic moiety. One particularly useful quaternary ammonium
compound, N-methyl N,N,N-tri(n-octyl)ammonium ion, used in the examples
herein, has alkyl groups with a straight chain length of one carbon for one R
group and a chain length of eight for the other three R groups. The R groups
may be the same or different and be aromatic, aliphatic, or mixed
aromatic/aliphatic. The R groups can contain other groups or other atoms of
Si, F, Cl, Br, 0, N, S, or P so long as they do not make the ionic compound
too oil insoluble, too water soluble, or neutralize or change its electronic
charge. Therefore, halogens such as chlorine and the like, halogenoids,
ethers, esters, imines, ketones, phosphate esters, nitriles, and the like are
permissible.
The extraction phase to aqueous phase ratio (E/A) is typically of about
1:20 to about 20:1 in the extraction circuit, preferably 2:1 to 1:10 and more
preferably 1:3 to 1:7, and most preferably 1:4 to 1:6. The extractant
concentration in the E-phase needs to be at least 0.1% and can be neat
(100%), more preferably 2-30% and most preferably 3-15%.
The pH of acid mine water is typically about 4-7 while in the mine and
not exposed to air. On seeping or flowing from the mine, or otherwise
exposed to air, the pH typically drops to about 2 to 4 depending on the
dissolved metals in the water, especially when ferrous ion, ferric ion, and
aluminum ion are present in the AMD water, normally the case.
The invention provides efficient means to purify water, especially the
removal of anions and cations present in the feed water contributing to the
waters' total dissolved solids level, collectively referred to as total
dissolved
solids solutes, values and/or contaminants. Though applicable for purifying
water at all scales, most preferably the invention is useful in purifying very
large volumes of water of tens to ten thousand gallons per minute flow rate in
a continuous-flow fashion using a unique combination of fast kinetics process
chemistry that makes use of interacting thermodynamic driving forces of ion
pairing, acid-base chemistry, gas evolution, and physical separation
19
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
technologies based on flotation (F) and liquid-liquid extraction (LLX)
technologies applied in liquid-liquid skimming configuration, and therefore,
is
referred to as flotation liquid-liquid extraction process technology.
Flotation
liquid-liquid extraction technology of the invention provides a major array of
s benefits including:
= Fast removal of water contaminants in seconds to minutes in total or
per treatment stage, rather than hours or days and weeks required by
conventional approaches.
= Fewer water contacting stages (where it is noted that, especially for
high flow rate feed waters, the larger the number of stages required to purify
the water, the more costly such treatment becomes since the same large
volume of water needs to be retreated at each stage),
= Removal of metal ions normally not extractable or not well extracted
by conventional liquid-liquid extraction (LLX) methods, and/or which foul
conventional LLX processes due to in-process solids precipitation, especially
Fe, Al, and Ni.
= Extraction of a broader spectrum of ions, anionic and cationic, than
conventional methods.
= Extraction of anions including sulfate, selenate, arsenate, molybdate,
vanadate, gold halo/pseudohalo complexes, chromate(VI), permanganate,
and the like, and cations including Fe(III), Fe(II), Cr(III), Cu(II/I), Ni,
Co(II),
Co(III), Zn, Al, Mn, Mg, Ca, La, Ga, Cd, Hg, CH3Hg+, Ag(I), Au(I&III) and the
like, to low residual concentrations rendering the water suitable for potable
water production, agricultural use, industrial process water, and the like.
= Concentration rather than dilution of contaminants with preferred
separation of the extracted ions into product concentrates, without being
bound by low value waste minerals, normally forming concentrates such as
metal ion sulfate solutions, solid salts, and/or solids of oxides, hydroxides,
bicarbonates, and/or carbonates, allowing them to be used to make useful
and saleable conventional products such as metal production (iron, zinc,
cobalt, nickel, copper, aluminum, and the like), sulfate-based fertilizers,
especially potassium sulfate, ammonium sulfate, soil acidulants, and/or
CA 02911119 2015-11-03
= WO 2008/100610
PCT/US2008/002092
sodium sulfate, a unique composition of matter consisting of a combination of
ferric, ferrous, or aluminum sulfates, and the like, with many applications.
= Low-cost means to purify large volumes of water continuously, in a
simple manner, with a high yield of purified water, and with a relatively
small
equipment size when compared to conventional technologies.
= Concentration of cations and anions in one or more liquid-liquid
extraction stages without formation of solid product waste sludges such as
precipitants, gypsum, lime sludges, or the like.
Description of Acid Mine Waste Drainage Water purification process without
waste generation and with metals and sulfate product (values) recovery.
The invention includes a second new composition of matter material
consisting of a unique formulation containing at least one liquid extractant
component combined with a metal ion colloid consisting of one metal ion in
neutral combination with anions of oxide, hydroxide, carbonate, sulfate
and/or bicarbonate ions that, when combined, form a colloid, floc, or
suspended particulate, that is useful in the simultaneous and rapid separation
of metal ions and anions from very large volumes of water at low cost and
without waste generation.
The water insoluble floc produced in the extractors of the F-LLX process of
the invention in the pH range of 5 to 8 is known to consist of the following:
= metal ions derived from the AMD water contaminants, especially Fe
and Al,
= cationic extractant derived from the extractant phase,
= hydroxide ion derived from the reaction of carbonate ion with water
during the extraction operation, and
= sulfate ion removed from the AMD water by the extractant phase.
This brown floc is less dense than water despite it containing inorganic
components. It can be isolated by filtering or preferably by centrifugation
after separating it from the two phase contents of the settlers and decanters.
The ElD decanter is the primary apparatus and operation to prepare this new
floc material since the floc generated there is in largest quantity and
contains
21
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
the least water and diluent. The floc solid is useful in purifying water by
enabling removal of TDS from contaminated waters and as a flexible means
for preparing metal ion salts and alkali salts of sulfate ion but without the
introduction of other salts that is common to these synthesis methods since,
with the floc, the counter ion is the extractant. Although carbonate ion
loaded extractant phase is used to raise the pH of the AMD water, as an
example, carbonate ion itself is not a component of the oil soluble floc
previously described, rather it reacts with water by hydrolysis to generate
hydroxide ions, that forms metal hydroxide colloids (see below), and it forms
certain insoluble metal ion carbonate particulates that are not part of the
floc
(see below). Hence, since the metal ions and sulfate ions are removed from
the water phase when the floc is formed at pH 5 to 9, the floc empirical
formula is estimated to be,
(R4N1x(N(II))40H-)z(S042-)w (for the case of divalent metal ions, N) (Eq. A)
or
(R4N+)x(M(III))v(OH-)7(S042)w (for the case of trivalent metal ions, M) (Eq.
B)
(Where M can be Fe(III) or Al, and N can be Fe(II), Ni, Co(II),
Cu, Zn, Pb, Cd, or Mn)
Now, using charge balance, the following relationships hold,
x n=z w, for the M case ...(c)
x+y=z+w, for the N case ...(d)
AMD water analytical results provided in Table 2A were used with the
above observed descriptive and chemical analyses to determine the values of
x, n, y, zand w. These results are given, using the Fe(II) and Fe(III) cases,
as follows:
Floc Chemical Composition: (R.4114)x(Fe2+)y(OHI(S042-)w
For which, x= 2 ¨ 3, y= 1 - 0, z= 2 and w = 1-1
22
CA 02911119 2015-11-03
= WO 2008/100610
PCT/US2008/002092
And
Floc Chemical Composition: (R4N+),(Fe3+)y(OFI-MS042-)w
x= 2 ¨ 3, y= 1 - 2, z = 2 and w= 1 - 2
Results in Table 28 generated the values of x, y, z, and wfor the following
extraction phase as follow:
(R4N+),,(Fe2+)n(OFIUS042-)w
x=2-4,n=1-0,z=2andw=1-3
(R4N+)x(Fe3+)y(OH):(5042-)w
x=2-4,y=1-2,z=2andw=1-3
For highest yields of these materials, it is important that the quaternary
ammonium ion is used in excess or at least in charge balance as calculated
using the above equations and for the AMD water composition of cations.
This material is insoluble in water and is oil phase dispersable, it can
contain solvent molecules of water and/or alcohols and/or esters of carbon
number of at least eight, preferably 8 to 24, and most preferably 10 to 16, so
long as the quantities of solvent do not collapse the colloidal material or
render in water soluble. The invention is a new composition of matter useful
zo for the low-cost purification of acid mine or acid rock drainage or
ground
waters, natural gas well brines, extractive metallurgical aqueous processing
waters, surface finishing process waters, agricultrual processing and waste
waters, and the like. It is especially useful to reduce high metal ion and
sulfate ion concentrations in water, for example as is found in mining and
mineral processing effluent waters, in brackish and brine waters, in
industrial
process effluent waters, livestock farm waters, and the like, down from very
high levels, for example total dissolved solids values of 300-5000 mg/L, down
to levels that allow water re-use, for example to < 500 mg/L total dissolved
solids for surface water discharge, or < 250 ppm to enable re-use as
drinking/potable water, and/or to deionized water with total dissolved solids
of < 125 ppm in which most or all of the contained toxic metal ions are
removed to < 1 or normally < 0.3 ppm, and sulfate ion to < 30 ppm,
23
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
phosphate ion to < 10 pppm, and nitrate ion to < 10 ppm. It is believed that
this efficient deionization is accomplished by a unique physio-chemical
process based formation of a new hydrophobic colloidal material consisting of
a oil soluble quarternary ammonium compound or blend of such compounds
(and/or a oil soluble phosphonium compound or blend of such compounds)
combined with a blend with of at leaste one, and preferably more than one
type of metal ions with anions selected from the list of oxide, carbonate
bicarbonate, hydroxide ions, sulfate ion, phosphate ion, and/or nitrate ions,
and where the colloidal material can be in suspended precipitate form, but
most preferrably it is in floc form, and most preferrably in colloidal form.
The
colloid, floc, or suspended precipitate can optionally also contain up to 70%
entrained water, preferrably less than 25% water, and more preferrably <
10% water, and most preferrable <7% water. In addition, the colloid, floc, or
suspended particle and/or precipitate can also contain up to 80% oil soluble
ts alcohol, ester, alkyl phenol, and/or other modifier, and most preferably
also
contains a diluent. All of the organic extractant phase materials are prefered
to be nonflammable compounds.
As is known in the art, sulfate ion is very difficult to remove from water
regardless of purification technology selected for the important niche of
medium to high flow rates when present at medium to high levels of SO4= in
the range of about 500 to 10,000 mg/L. The need is to reduce the sulfate ion
concentration far enough to enable surface discharge release of the water,
into the range of 0 to < 600 ppm needed for surface discharge release, and
preferably to enable its use for potable water (< 250 ppm), and most
desirable to < 100 ppm needed to prevent drinking water "sulfur" odors. This
difficulty in removing sulfate ion is well known in the art of water
purification
and is due to sulfate ion's high charge density and high water solubility of
all
its salts except barium ion, and barium ion is very toxic, very expensive,
very
difficult to recycle, and is in severely limited supply relative to the amount
of
water needed to be purified with respect to ,sulfate ion. The commercially
available technologies all have the a major problem in that a substantial
amount, normally 25 to 50%, of the water to be processed ends up as a
24
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
waste stream carrying the contaminants from the process to regenerate the
sorbent or avoiding scaling of the process hardware. Hence the technologies
of the known art produce a large waste stream that is still of a very large
volume, still dilute, still toxic, and still represents a severe and large
disposal
problem.
In the present invention, a unique "forced" ion pairing process
chemistry is used to quickly remove highly soluble and/or highly charged
anions, especially SO4= to < 100 mg/L residuals, and in high yield from these
intermediately concentrated (about 600 to 10,000 mg/L total dissolved solids
lo feedwaters. The invention maximizes the use of off-the-shelf hardware to
help speed up the availability and widespread use of this much needed
technology.
The process chemistry of the invention involves contacting (mixing)
contaminated aqueous solutions with a specially formulated, nonflammable,
water immiscible, oil soluble, extractant phase. The sulfate is extracted into
this second liquid phase, that is also used at a volume (or flow rate) that is
substantially less than that of the contaminated feedwater so that the sulfate
and other ions co-extracted are pre-concentrated by the factor of the volume
(flow rate) ratio. In this manner the water impurities are not only removed
from the water but are simultaneously concentrated multiple times so that a
concentrated metal sulfate product of saleable value is produced rather than
production of wastes. To boost process efficiency and to lower process costs,
an aspect of the invention is that, at the same time the sulfate ion is
extracted, the toxic metal ions contained in the feed water are also removed
and concentrated using the same operation. The capability of the extractant
phase to extract both sulfate ions and metal ions simultaneously into a water
immiscible phase is key to the invention and occurs due to the process
chemistry and process operating conditions selected for the invention.
This colloid/floc process chemistry mentioned above is described
further below.
The extractant chemistry discovered for the role of SO4 removal is a
formulation containing an oil soluble quaternary amine {R4N+). The use of
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
quaternary ammonium LLX extractants for anion extraction is known in the
prior art. However, this art has consistently indicated that sulfate ion is
only
poorly extracted by these reagents and far prefers extracting mono-anions
and anionic metal ion complexes instead. Therefore it was necessary to
s modify the process chemistry such to promote sulfate ion partitioning
into the
water immiscible extractant phase. This "forced" approach to the process
chemistry will now be described.
Choice of Anion and Base for pH Control
Since acid mine drainage waters and the like are already very highly
excessive in total dissolved solids level, simple ion exchange removal of
sulfate ion, for example by using the chloride ion form of the quaternary
ammonium extractant, was out of the question since any anion exchanged for
the sulfate ion would then render the water still a waste too toxic for
discharge to surface waters, much less be of any value for use by industry or
residential. Therefore anions that can be eliminated by forming H20 and/or a
gas, specifically CO2 gas, were used to develop the process chemistry, that is
the hydroxide ion (OK), carbonate ion (CO3'), carbonic acid (H2CO3), and/or
bicarbonate ion (HCO3-) form of the quaternary ammonium extractant). It is
also important that the quaternary amine does not require protonation to
possess a positive charge and is positively charged over the entire pH range.
In this manner the strong base options of carbonate ion and/or hydroxide ion
can be used. The process chemistry identification and selection is further
developed below to explain the fundamental separation process chemistry the
proposed mechanism of action.
Preferred Extractant Structure and Means of Process pH Control
A quaternary amine liquid-liquid extractant particularly suitable to
practice this invention is N-methyl tri(octanyl) ammonium ion available as
Aliquate134 or Aliquat 336 (Cognis, Inc.), or oil soluble, low water soluble
quaternary ammonium compound, preferably a liquid compound, and most
26
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
preferably a branched or extensive tripodal structure to discourage gelation
and solidification throughout the process load, strip and storage cycle.
Description of Colloid Formation and Its Impact on Separation Process
s Chemistry Basis
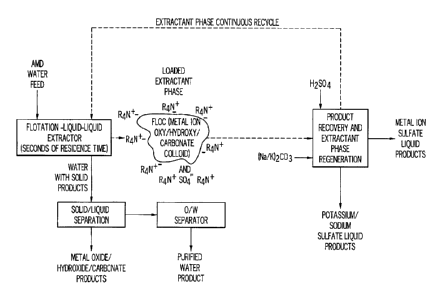
One embodiment describing the invention is the general process flow
diagram Figure 1A using acid mine drainage water as the "Metal Sulfate
Laden Feed Water" 1110 example for the description. The first step involves
treatment of the acid mine drainage water stream with a particular Aliquat
io 134 ¨ based extractant phase formulation 1010 in a self-controlled pH
buffer
system of carbonate/bicarbonate buffer, included in the extractant phase
formulation with the Aliquat 134, and controlled in the preferred range of pH
5.5 and 9.7 for at least one stage, preferably two stages, and where up to
five stages is effective and 4 stages with the preferred pH range for
extraction
is stage 2030 ranges from 7,4 to 8.2 and is preferably 7.6 to 8.0 and is
most
preferably about 7.8. The stages can be configured entirely counter-current,
or cross-current where one or more sulfate-stripped extractant phases (see
below) are blended with the acid mine drainage water as it flows serially
through two or more stages, or co-current flow, cross-flow, or a combination
20 of these flow configurations. Most preferred is to operate the
extractors with
the four stages separated into two sets of at least two mixer settlers each,
where each set is arranged counter-current with its partner, and each set
internally is piped up to be counter-current, but the two sets are configured
cross-current flow with respect to each other where the extractant phase is
25 the crossing phase and the aqueous phase is piped to flow serially from
stage
to stage across the first set and then across the second set, and then, if
present, across the third set, and so on. However, other combinations are
easily arranged and tested since piping changes are readily made as is
already well appreciated in the current art of liquid-liquid extraction
30 technology using staged devices such as mixer-settlers, centrifuges,
columns,
in-line static mixers, and the like and any combination of these. Most
preferred is the, in this case is a combination of counter-current and cross-
27
CA 02911119 2015-11-03
' WO 2008/100610
PCT/1JS2008/002092
flow configurations, described above, while operating the strippers always
with counter-current flow and internally aqueous recycled. Also, most
preferred to the invention, is that the extractant phase approaches in the
settlers are specially designed for floc handling by promoting simultaneous
floc, colloid, floating precipitate, extractant liquid phase, and/or gelling
mass
densiflcation, while still maintaining its continuous, or at least
intermittent,
flow and by promoting the flow, at least intermittent and preferably
continuous, of this floc over the extractant phase exit weir, by any
combination of mechanical movement and/or smooth, curved surface as, for
example, is found in ore flotation cell designs, and lastly by accommodating
the flow requirements of this extractant phase "floc", preferably thickened,
to
the next stage, where the flow can be intermittent or continuous, whether it
be another extractor stage, a decanter for further dewatering, or a stripper.
The receiving hardware for the floc from the flotation overflow weir can be of
hopper design to send the floc to the next stage, a pipe of preferably
relatively large ID, or a trough, a hydrocyclone, or any combination of these.
The most preferred case is that the following stripper uses an aqueous
acid to dissolve the components of the extractint-based floc, whether it be a
colloid, precipitate, slurry, liquid, emulsion, gelling mass, or any
combination
zo of these, thereby returning the system back into a liquid-only
condition.
Liquid-only systems are easiest to handle and process under continuous flow.
Suogested Mechanism of Action
While not wishing to be bound by theory, but acknowledging that
theories are useful in understanding physical/chemical processes, the
interrelationships of control parameters with process outputs, capabilities of
the process, and the like, it is presently believed that contacting
contaminated
water, preferably containing dissolved minerals of cations and anions, with
the invention extractant phase method of operation, and hardware generates
a hydrophobic and variable combination of oxo and hydroxo, optionally in
combination with carbonato, bicarbonato, sulfato and/or other metal on
ligand components and anions present, with metal ions containing
28
CA 02911119 2015-11-03
= WO 2008/100610
PCT/US2008/002092
homogenous, but usually heterogenous, material with the physical
consistency, depending on age, temperature and concentration, with the
properties of one or more of the following: metal ion complexes, clusters,
colloids, flocculated particulates, floating precipitates, liquids and/or
gels, and
that can have various degrees of hydration from low to high, and will be
collectively referred to as a "floc" in this description. Microscopically, it
is
believed that this floc is essentially neutral but preferably slightly anionic
so
that it interacts with a positively charged hydrophobic "extractant" dissolved
in a water immiscible liquid. At the molecular level, it is believed that this
floc
forms quickly, within a few seconds since metal ion to ligand bonds need not
be broken or formed. It is believed that this capability is an important
feature
that makes the invention immensely useful for cheaply purifying large water
flows of a broad spectrum of contaminants. The floc consists of oxo,
hydroxo, with or without other anions, providing colloid forming complexes of
the metal ions present, for acid mine drainage waters normally one or more
of Fe(II), Fe(III), Al, Mn(II), Mn(IV), Ni, Co(II), Co(III), Zn, Mg, Cu, and
Ca.
As provided by the inventive formulation, simultaneously sulfated quaternary
amine, (R4N)2SO4, also forms and removes sulfate ion from the water in high
yield, and, in another aspect of the inventive formulation, simultaneously
neutralizing the acid present in the acid mine drainage water by the
conversion of H+ ions present and provided by acidic metal ions into H20 and
H2CO3 molecules by neutralization with CO3 and/or HCO3- ions to make
H2CO3, and, in another aspect of the inventive formulation, simultaneously
reducing the total dissolved solids level of the water through the
dissociation
of H2CO3 into CO2 gas and H20, thereby producing a purified aqueous phase
(water). Selenate and silica/silicate are also recovered. The metals
recoverable include those capable of metal hydroxide precipitations, metal
carbonate solid formation, metal sulfate solid formation, or those metals that
co-precipitate by inclusion in to ferric hydroxide and/or aluminum hydroxide
amorphous precipitates, and/or those anions that form highly insoluble
precipitates with ferric ion or aluminum ion, such as arsenate, phosphates,
humic acids, other natural organic matter, and the like. This includes all the
29
CA 02911119 2015-11-03
* WO 2008/100610
PCT/US2008/002092
metal ions on the Periodic Table except alkali metal ions, which are reduced
only a low to small amount. Alkali metal ions are typically a minor component
of ground waters, surface waters, farm waters, and most industrial process
waters. The low removal of alkali metal ions is not a detriment since in fact
there is much allowance by water users that these ions can remain in the
product purified water, especially the nutrient r and where softened water is
used (where Na + residuals exist in the treated water). For this reason also,
the technology of the invention is not useful for the desalination of sea
water
where the main objective is the removal of high concentrations of the alkali
salt NaCI is required. ,
Metal ions removed by the process typically constitute those metals
that form oxide, hydroxide, or carbonate colloids, and those that co-
precipitate with such materials (Figure 2). Also attached is Table 3
summarizing the list of effective extractant structures. These can be used in
water immiscible solution form. They can be used neat if their corresponding
bicarbonate/carbonate/bisulfate or sulfate ion forms are sufficiently fluid at
short mixing and settling times (see below) so to enable flow in batch or
continuous liquid-liquid separation process or flotation process or oil
skimming
circuits. Note that the prior art teaches that all such metal hydroxide and
insoluble salt composition systems will form solids which precipitate within
conventional LLX hardware requiring process hardware shutdown, solids
removal, repair of damaged equipment and other maintenance. The current
invention avoids this serious problem. If enhanced fluidity is needed as is
normally the case, then one or more of the quaternary extraction compounds
are blended with a predominantly hydrocarbon diluent of eight or more
carbon atoms. Suitable diluents are included in Table 3. Also included in
Table 3 are candidate "modifiers" that can be added to the extraction
formulation that can aid in displacing entrained water in the extractant phase
s carrying the sulfate ion and dispersible metal ion complexes and/or colloids
of
oxo, hydroxo, carbonato, and/or sulfato extractant phase flocs. Modifiers can
also help the floc to solubilize in the hydrocarbon diluent.
CA 02911119 2015-11-03
= WO
2008/100610 PCT/IIS2008/002092
Incorporation of Floc Filtration Option
In another embodiment of the invention, the metal ion/sulfate ion floc
material, loaded onto the extractant phase during the mixing step (Figure 1C)
is filtered after the extraction operation and/or in between extraction
stages,
.. to remove the novel composition (described above) consisting of the
formulas:
(for the case of divalent metal ions, N) ...(a)
and
(R4N+).(M(III))v(OH-)2(S042-)w (for the case of trivalent metal ions, M)
...(b)
This filtration illustrates that the new material described can be produced as
an isolated product. This filter step is optional and would be performed
before
the metal ion and sulfate ion stripping steps (see below). The filtrate, now
essentially 100% liquid colloidal floc and liquid, is then processed using
liquid-liquid contactors in the stripping of soluble metal ions and of sulfate
as
described below.
Sulfate Ion Product Production with Concomitant Regeneration of Carbonate
.. Form of the Extractant Phase
After liquid-liquid stripping and/or filtering the metal ion products from
the extractant phase, as described above and in the attachments and
examples below, the essentially metal ion¨free extractant phase still contains
the sulfate ion (and any selenate ion) recovered from the acid mine drainage
feed water during the extraction operation. The chemical form of the sulfate
ion on the extractant phase is believed to be as ionic compounds (RN)2SO4
and/or R4N+HSO4- depending on the pH of the extraction or metal ion
stripping stage operation stage from whence in came, normally the former
from the extraction circuit and the latter from the acidic metal ion strip
circuit,
is contacted in counter-current LLX configuration with 0.1-55.% Na2CO3or 0.1
¨ 55% K2CO3 solution, or solutions of bicarbonate ion, or blends of these,
with or without added NaOH or KOH up to saturated conditions and could be
31
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
optionally warmed to achieve even higher extractant phase loadings of these
anions. Total concentrations of carbonates than solubility limits can be used
provided the resultant slurries are kept in flow motion, at least
intermittently,
by mechanical means of stirrers and pumps and use of troughs and large
piping instead of small ID piping typical of conventional LLX technology. With
use of suitable solids handling equipment, i.e. hopper with auger addition
trough, solid carbonate and hydroxide solids can be added directly to the
sulfate strip mixers as powders, pellets, granules, and the like. With this
information, it would be obvious to others skilled in the art of conveying
1.0 slurries to prepare related mechanical designs for the physical
handling of the
product "floc" of the invention as described above.
At the preferred counter-current arrangement of the sulfate stripper
operation the "first" sulfate strip stage, "S1-504", generates an aqueous
raffinate from the sulfate stripper stage that is the most concentrated in
sulfate ion and represents the "sulfate product concentrate". Depending on
relative flow rates of the extractant phase and the carbonate/hydroxide strip
feed solution, the sulfate ion product concentration can be adjusted over a
very wide range of approximately 2,000-650,000 mgS047L. Preferably the
product sulfate ion concentrate is 150,000-250,000 mg/L (or about 15-25%)
for the case of stripping with Na2CO3, and 5,000-150,000 mgS047L for the
case of stripping with K2CO3.
Although in the prior art the anion-anion liquid-liquid exchange
involving sulfate ion is normally achieved with poor efficiency and
incompletion in the case of extracting sulfate ion from water, the nature of
the invented process disclosed herein provides sharp, fast and high yield
sulfate ion recovery and stripping in stages Sy-SO4 (where y = 1 to 3 stages,
preferably 1 to 4 stages, and most preferably 1 to 5 stages, while 1 to 6
stages is also effective), by providing the combination of strong
thermodynamic driving forces of acid/base neutralization {favorable
(negative) enthalpy change} and innocuous gas (CO2) formation {favorable
(positive) entropy change}, and low E/A ratio, dianion exchange, and release
(positive mass action effects), summarized by the following equations for
32
CA 02911119 2015-11-03
. WO 2008/100610
PCT/US2008/002092
sulfate ion (equivalent reactions can be written for nitrate, chloride,
methane
or others sulfonate(s), phosphate, acetate, and other anions that represent
the conjugate base of the acid used in the metal ion strip circuit used
separately or in any combination), and where Extractant Phase = E-phase,
s and the undesignated phase is Aqueous Phase,
2 {R4N+HSO4-}E-priase + MxCO3 4
H20 + CO2(9) + {(1141+)2SO4=}E-Phase + MxSO4 (1)
For example, for x=2 and 504= strip stage pH of ¨2 < pH < ¨7,
2 IR4N+HSO4-1E-Phase + K2CO3 4
H20 + CO2(9) + {(R4N+)2SO4=} E-Phase + K2SO4 (1')
and/or,
2 {R4N+HSO4-}E- phase + 2MxCO3 4
2M(HCO3) x + {(R4N+)2SO4'}E-phase + MxSO4 (2)
For example for x=2 and SO4= strip stage pH of ¨8.5 < pH < -40,
2 {R4N+HSO4}E- phase + 2K2CO3 4
2KHCO3 + {(R4N+)2SO4.1E-phase + K2SO4 (2')
and/or
{(R4N)2SO4}E-phase + MxCO3 4 {(R4N)2CO3)E-phase + MxSO4 (3)
As an example, for x=2 and SO4= strip stage pH of > 10,
{(R4N)2SO4}E-phase + K2CO3 3 {(R4N)2CO3}E-phase + K2SO4 (3')
and/or
{(R4N)2SO4}E-phase + 2M(HCO3) x 4 2{R4NHCO3)E-phase + MxSO4 (4)
Where x is 2, and where the {R4N+HS0.4}E-Phase and/or {(R4N)2SO4}E-phase
species flow into the sulfate ion strip circuit from the metal ion strip
circuit(s)
and the anion is that of the strip acid used in the metal sulfate stripping,
and
33
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
for acid mine drainage water feed may also contain sulfate/bisulfate ion. The
relative contribution of each of the above four reactions at each stage of
stripping depends upon the pH of operation of the last stage of the metal ion
stripper circuit (or of optional wash stage(s)), that is, whether the anion on
s the extractant phase is sulfate ion, bisulfate ion, or a blend of these,
and on
the pH of each stage of the sulfate strip circuit. The use of pH to control
the
strip circuits, including more specific effective pH values, is further
detailed
below.
For the above, M is most preferably Na + or r (x=2), or blends
thereof. These ions can be used alone or combined with any of the following:
NH4 + (x=2) provided the pH is < ¨9) and especially as concentrated solutions
of ammonium bicarbonate (NH4FIC03) to produce concentrated product
solutions of ammonium sulfate, ammonium nitrate, ammonium phosphate,
and the like and/or blends thereof. Given the above information, it is obvious
to those skilled in the art of liquid-liquid extraction that the basic anion
can
also be added back onto the extractant phase after the stripping of the
sulfate
(or other anion) by a neutral anion, e.g. sodium or potassium nitrate and/or
phosphate solution, followed by a separate sequence of contactors to replace
these ions with carbonate ion, or the like. This mode of operation however
requires additional contactors and chemical raw materials, and is therefore
less preferred.
The generation of CO2 gas in this manner is a unique feature of the
invention and occurs most when the last stage of metal sulfate stripping (S3M
in the most preferred case), or wash stage (also in the most preferred case)
operations are acidic (pH<3, and most preferably pH< 1), as it is in the case
where the strip acid conjugate base is a weak acid, as it is in the case of
sulfuric acid (bisulfate conjugate base), orthophosphoric acid
(orthophosphate, monobasic being the conjugate base). It does not occur
significantly when the conjugate base of the strip acid is not a weak acid,
for
example when nitric acid or hydrochloric, other hydrohalic, MSA (methane
sulfonic acid), and the like.
34
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
The CO2 release occurs preferentially in stage the first stage of anion
stripping. For the sulfate case this is stage S1-SO4 (first stage of
carbonate/bicarbonate stripping of bisulfate ion) and is due to Reaction 1
where the control pH at the S1-SO4 mixer is most preferably 4.5 1,
preferably 4.5 2, but control in the pH window of 2 to 7 is still effective.
The
CO2 so released is humid, but otherwise pure if the S1-SO4 mixer is
reasonably sealed against air intrusion. This CO2 gas is ideal for adjusting
the
pH of the finished water back down to <9 when the final extraction stage is
operated at pH 9-11 to remove Mg and Ca to soften the product water and
to further reduce total dissolved solids (see below). Being pure, the CO2 gas
can be captured, pressurized and packaged as liquid or dry ice by current
commercial and well established technology to produce a useable CO2
product. Otherwise it can be harmlessly vented. This CO2 gas does produce
a rapidly breaking foam in the S1-504 mixer that needs to be handled such to
prevent tank overflow spillage during the operation. If the E/A ratio in the
stripper is insufficient in phase, that is the pH in S1-SO4 stage exceeds ¨7,
then some foaming may aqueous also occur in stage S2-SO4. This foaming is
found to be a minor issue as the foam breaks rapidly and only a small foam
head is produced. For this reason a continuous stirred mixer tank is most
preferred with higher walls on the mixer and associated settler compartment
of the first and second carbonate strip stages compared to conventional
liquid-liquid technology (see below). Most preferred is that S1-SO4" also
contains a cover that enables the capture of the CO2 gas, without interfering
with the mixer, to use to sparge through the purified product water (see
below), if needed, to optionally adjust its pH downwards, preferably to pH <
10, and most preferably to pH 5_ 9.
When the aqueous carbonate solution being flowed to the S1-SO4
sulfate stripper stage is derived from a sulfate ion stripper stage just up
stream of the S1-SO4 stage in a counter-current operation, i.e. S2-SO4, the
most preferred case, then some or most of the carbonate may already be in
the bicarbonate ion form (see above chemical equations), i.e. MyHCO3, where
y = 1 for M = Li+, Nat, K+ and/or NH4, and y = 0.5 for M = Mg2+, Ca2+
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
and/or Zn2+. If M=NH4+, as is well known in the art of handling ammonia
aqueous solutions, that the form of a very soluble reagent addition is
NH4HCO3, prepared from a solid and/or concentrated solution, or directly from
NH3(g) gas or aqueous ammoniacal solution, NH3(aq).
The extractant phase from S1-SO4 can still contain about one half of
the sulfate ion from the extraction circuit depending upon the overall E/A
ratio
of the stripper circuit and the concentration of active quaternary ammonium
concentration used in the extractant, and the E/A flow ratio used in the
extraction circuit. This phase is then contacted, preferably in counter-
current
liquid-liquid extraction mode using suitable hardware (e.g. mixer-settlers,
static in-line mixers, columns, or continuous liquid-liquid centrifuges, or
hydrocyclones), at least one additional contact time, preferably two to three
more, and most preferably four to six more times; that is to say using
additional counter-current configured stages, for example S2-SO4, S3-SO4,
and S4-SO4 for a total of four anion (sulfate) strip stages linked counter-
current. Hence the extractant phase exiting the "last stage" of the sulfate
ion
strip circuit (normally S4-SO4, but could be S3-SO4, S5-SO4 or S6-504), is the
carbonate ion-loaded extractant phase (i.e. the form containing
{(R4N+)2CO3-1
JE-phase) that is recycled to the extraction circuit (e.g. see
attached detailed process flow diagrams).
If hydroxide ion, OK, is to be also added to the CO3'-loaded E-phase
to reduce the amount of residual total dissolved solids by forming less HCO3-
in the purified water and/or by removing hardness metal ions, then OH- is
added to the SO4= strip stage mixer (normally to stages S3-SO4, S4-SO4, S5-
SO4, and/or S6-SO4 depending on the desired (KW) residual desired in the
treated water and/or on the E/A used in the anion stripper operation).
The raffinate phase flowing from the S1-SO4 stage, either continuously
or intermittently, is the useable sulfate ion product, for example
concentrated
solutions and/or easily crystallized solids of (NH4)2SO4, K2SO4, and/or Na2SO4
(as a hydrate or anhydrous form). These products are in addition to the
metal salt solution products or solids, for example metal sulfates produced
from the S1-M metal ion strip stage circuit referred to above, or from both S1-
36
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
M and S1-N stages from a dual metal ion strip circuit (see below). These
products are items of commerce used in numerous industries, with the metal
salt concentrate blend being of a new composition (see above).
As illustrated by the discussion above, and the Detailed Description of
.. the Invention and Examples sections, the exact pH and E/A flow ratios of
each flotation liquid-liquid extraction contactor, the added reagents and
reagent concentrations, and the like, used determine the purity of the water
produced and the type and amount of co-products produced. This flexibility
of products help insure that the product mix and purities produced can be
best tuned to meet regional municipal, industrial, agricultural, and
residential
market demands for the products produced in practicing the invention.
Operation of the Sulfate Recovery Circuit and Regeneration of Carbonate form
of the Extractant Phase for Continuous Recycle
In a most preferred embodiment, if the feed of acidic extractant phase
to S1-SO4 is in excess of the molar amount that can be handled by the above
chemical reactions 1 to 4, then all of the aqueous carbonate is consumed, as
desired, in S1-SO4, as given in the above chemical equation, thus insuring
that no carbonate exits the process with the sulfate product and thereby
insuring that all the carbonate added to the system is transferred to making
sulfate concentrate product (or other anion product as listed above). In the
case of some excess acid being present in S1-SO4 (indicated by a S1-SO4
mixer or raffinate pH of <4), (the acidity being supplied from the last metal
ion stripper stage, normally S3M, but optionally could be S2M or S4M), and
present as {R4N+HSO4}1-phase, or as {R4N+H2PO4-}E-phase and/or {R4N+HPO4=}E-
phase if phosphoric acid is the strip acid used, still exists on the
extractant
phase and this is easily consumed, and the associated sulfate removed, in the
S2-SO4 stage, but CO2(g) does not form, as follows (shown for the sulfate ion
system):
{R4N+HSO4 ¨}E-phase + MxCO3 4 M(HCO3) z + {(R4N+)2504= }E-phase
(5).
37
CA 02911119 2015-11-03
= WO
2008/100610 PCTAIS2008/002092
and in parallel,
{R4N+HSO4 -}E-phase + MxCO3 MxSO4 + 2{R4N+ 1-1CO3-}E-phase
(6)
Where the M, x, y and z definitions are as before. In this case, the
extractant phase carries the {(R4N+)2SO4 =}E-phase and {R4N+ HCO3-1
, E-phase
species to the S3-SO4 and/or S4-SO4 strip stages where the SO4= and HCO3-
extractant phase species are displaced (ion exchanged out) by the
overwhelming high concentration of divalent (pH 10+) CO3= ion. The
carbonate ion concentration used is limited only by the solubility of the
carbonate salt and, more preferably, also by the product of the salt solution
in
stage S1-SO4 (again, using the sulfate ion as the example for illustration
purposes). Hence when using potassium carbonate as the strip aqueous
solution, the effective concentration range is 0.1% to 50% K2CO3 at ambient
temperatures; but preferably 12-35 % K2CO3 so that a concentrated
potassium salt is produced from S1-SO4 stage that is supersaturated with
respect to K2SO4 crystallization, making recovery of a solid K2SO4 product,
with recycle of supernatant/filtrate preferred; and still more preferably 6-
11%
zo K2CO3 so that the salt produced in stage S1-SO4 remains soluble so that
a
liquid product that is readily saleable as liquid fertilizer is made, and so
to
avoid the potential for hard scale to form in S1-SO4 unit when the product
produced there is K2SO4, and most preferably 7.5 to 8.5% K2CO3 so that the
maximum (about 10-11%) concentration of K2SO4 solution, that does not
crystallize at room temperature, is produced.
Similarly, when using sodium carbonate as the strip aqueous solution
being fed to the last strip stage, and optionally, although less desirably, to
one or more of the others, the effective concentration range is 0.1% to 25%
Na2CO3 (as anhydrous) at about 25 C temperature, and greater at higher
temperatures, but preferably 6-15% Na2CO3 so that a concentrated sodium
salt of up to about 22% Na2SO4 is produced (as Na2SO4*10H20 if T is
<-33 C, and anhydrous if T is > -33 C) from S1-SO4 stage, and still more
38
CA 02911119 2015-11-03
' WO 2008/100610
PCT/US2008/002092
preferably about 10-15% Na2CO3 so that the salt produced in stage S1-504 is
soluble and sodium sulfate hydrate solids do not develop in S1-504 unit when
the product produced there is Na2SO4 solution.
The SO4= and HCO3" ions so stripped by the carbonate ion using the
solutions defined above then accompany the aqueous phase from the last
strip stage (normally S4-504, but can also be S3-SO4, S5-504 or S6-504
depending on the initial and residual (SO4') concentration objectives of the
feed and purified product waters of the extraction circuit), to 51-504 to
become part of the final products as given above. In chemical reaction form
these reactions for stages S3-504 and SO4 are
{(Ft4N+)2SO4 }E-phase M),CO3 MxSO4 + {(R4N+)2CO3=}E-phase
(7)
and/or,
2{114N+ HCO3 -}E-phase MxCO3 4 {(R4N+)2CO3-}E-phase + M(HCO3)z
(8)
Similarly for optional stages S5-504 and S6-504 if they are used.
zo Extractor Configuration to Maximize Separation of Trivalent Metal Ions
from
Divalent Metal Ions
Using the example of acid mine drainage or acid rock drainage feed
water, the partially purified acid mine drainage aqueous steam generated in
extraction circuit El (operating at pH 3-5 for trivalent metals) is directed
to
zs another extraction circuit (E2) for further processing at pH 5-10 (for
any
residual trivalent and especially divalent metals present in the water feed).
At
the lower pH, conversion of CO3= to CO2 occurs, thereby requiring the R4N+ to
ion pair with any remaining anionic, especially sulfate ion in the case of
acid
mine drainage, not extracted in El stage, and to also form additional
30 molecular metal ion complex clusters, colloids, flocs, particulates, and
the like,
containing hydrophobic quaternary moieties, as described above, with
hydroxides and oxides of metal ions, mostly ferrous ion in the case of acid
39
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
mine pool water, collectively illustrated as {(R4N),}{M(OH),}, along with
essentially separate and essentially soluble colloids of sulfate ion with the
quaternary ammonium ion extractant depicted as ({(R4N)2504}coiroid)v. For
these species "w" is the absolute number of charges per colloid, cluster,
particle, and the like, and also the absolute number of charges on the sulfate
ion colloids, to within a zeta potential of 25 mV or less.
To separate the lower pH extracting metal ions from those extracting
at higher pH, the extractant phase from above is then stripped with an acid
(e.g. sulfuric acid or one of the other acids listed above) to remove the
to trivalent metal ions, and any more acidic +2 metal ions such as
copper(II), as
metal sulfate concentrates, as described above, to then yield {R4N+HSO4-}E-
phase, depicted as {(R4N)2 SO4}E-phase. Trivalent and certain divalent metal
sulfates are recovered and are useful as chemical specialties and commodities
(see above). When the trivalent metals present are ferric ion and aluminum
ion, and also when the ferrous ion is co extracted and stripped with these
ions, then a unique ferric aluminum ferrous sulfate product aqueous
concentrate is so produced and represents a unique composition of matter of
value in large scale water purification applications that provide many
beneficial advantages over the separate components of ferric sulfate, ferrous
sulfate and alum. There are many other uses for this new composition of
matter product (listed above).
One new composition of matter was discovered and produced using
the invention and consists of a blend of Al3+, Fe3+, HSO4", and SO4- ions,
with
a second formulation of these ions and Fe2+ ions at concentrations and ratios
that maximize municipal and industrial waste water treatment, municipal
potable water, and/or industrial process water purification processes.
Conventional treatments involve the use of only one of the following for these
applications and at higher costs: ferric chloride, ferrous sulfate or alum.
However, although these reagents are functional in removing dissolved toxic
metal ions from water and coagulating suspended solids and biologically
derived biomass (from anaerobic digesters), they all have serious
shortcomings. First, though ferric ion is preferred for the above properties,
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
the chloride content of ferric chloride promotes corrosive ion water
distribution tankage and piping, leading to serious corrosion of copper
plumbing and to Pb-based solders in the water distribution system causing
serious birth defect negative health effects. Hence ferric chloride coagulate
has been reduced in use for potable water production. A second problem
with ferric chloride is that ferric ion too rapidly hydrolyzes to Fe0OH
precipitate, thus preventing it from dispersing well enough in the treated
water to enable its efficient use in removing other pollutants of concern,
especially arsenic As and phosphate ions (P043-). Alum (an aluminum sulfate
hydrate) is desirable in water treatment due to ease of dewatering but suffers
from the deficiencies of narrow operating pH range and especially in forming
excessively voluminous sludge, some ten times or more that formed by ferric
salts. Ferrous sulfate avoids the problem with the use of chloride ion and is
water soluble around neutral pH enabling it to be thoroughly dispersed so to
more effectively precipitate toxic contaminants such as As and P043.
However, ferrous ion is a poor coagulant and flocculent and must be air or
chemically oxidized to provide coagulation and flocculation, a slow or costly
process, respectively.
The new composition of matter now provided by this invention avoids
the above problems while maintaining all of their benefits. The metal sulfate
concentrate product is thus unique for purifying a broad spectrum of waters
for many needs. It does not contain the corrosive chloride ion, it does
contain ferric ion in abundance for fast coagulation and flocculation, it does
contain ferrous ion that provides a fully soluble form of iron to effectively
precipitate As and P043, and lastly, it does contain a low amount of aluminum
ion that is sufficient to both perform coagulation and flocculation with a
"filter
aid" effect on the result and sludge filtration and dewatering steps, and yet
the precipitate is dense due to the ferric content that collapses the
otherwise
voluminous AlOOH precipitate.
This new composition of matter is formulated Fem.Fen Al2(SO4)w=H20
and is provided in both aqueous solution and solid forms. The equivalent
ratios (dry conditions where q=0), based on charge balance, are,
41
CA 02911119 2015-11-03
" WO 2008/100610 PC
TMS2008/002092
(X X 3) + (y x 2) + (z x 3) = 2 (9)
As an example, for x=1, y=1 and z=1, then the value of "w" is
(3+2+3+)/2=w=4, provides the empirical formula of FeiliFeliAl(SO4)4 =qH20
with a nonhydrous formula weight of 523 g/mole. Another example, based
on acid mine drainage water feed is x=0.1, y=0.8 and z=0.1, giving a value
for w = (0.3+1.6+0.3)/2 = 1.1, or Fen10.3Fell1.6A10.3(504)1.1 for a formula
weight of 159 g/mole. Specifically, this new material consists of the range of
ratios: FelI10.03Fell.95A10.03(504)1.04 (FW of 155 g/mole), to Feln0.95
Fe110.03
A10.5(SO4)2.2(FW of 280 g/mole). The total composition of the aqueous
solution of the new material, Fenix Fe" y Alz(SO4)w , produced by the process
of
the invention from acid mine drainage feed water is (dry weight) at least
0.1%, and preferably 1 to 5 %, and most preferably 5 to 20%. All
percentages are by weight in aqueous solution. The solid material is
prepared by taking the metal sulfate concentrate from the process of the
invention and either cooling it to 0 to 5 C and allowing the material to
crystallize, or by drying the material using heat, evaporative cooling, or the
like, until sufficient water has been removed to enable crystallization of at
least a part of the originally dissolved, and preferably precipitating at
least
90% of the dissolved solids, where upon a substantially amorphous, granular,
air sensitive material is produced. Up to 50% of moisture is left in the solid
to
retain the materials reactivity and water solubility. Oven drying the
material,
especially in the air or other source of oxygen gas, is not recommended since
at least a portion of the material then would form amorphous mixed oxides of
iron and aluminum, a portion would darken to magnetite (mixed ferric and
ferrous particulate) would be produced, and the sulfate would be converted
back to sulfuric acid making the material hazardous to handle. This new and
unique material is made by selecting a strip acid, specifically sulfuric acid
in
this case, that is used to strip the Fe(II), Fe(III) and sulfate ion
components
of a metal-loaded extraction concentrate at an E/A ratio such to provide
substantial concentration enhancement, at least 10 fold, preferably 50 fold,
and more preferably 100 fold. In addition to these parameters the product
concentration of this unique material produced is determined also by the
42
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
number of acid strip stages used and the number of internal recycles
achieved. The S1-M strip stage is the most influential on metal ion
concentrate. At the minimum, stripping must be sufficient to strip a
substantial portion of the metal ion content of the colloid loaded extractant
s phase while mixing for up to 30 min, preferably 15 min, and most
preferred
up to 6 minutes. Normally more than 99% of the metal ions of the colloidal
floc are stripped into the mixed metal sulfate concentrate due to the multi-
stage counter-current liquid-liquid stripper design of preferably three
contact
stages.
An important and unique provision of this invention is that, in one unit
operation with a relatively small size it provides:
1) Removal of toxic cations, including the very slow reacting ferric, ferrous,
nickel, and aluminum ions, and the like, even present at very high levels, to
levels compliant for surface water discharge, potable water, and other
regulated requirements,
2) Neutralization of the acidity that characterizes many waste streams,
including acid mine drainage, acid rock drainage, natural gas well, and the
like, discharge waters,
zo 3) Removal of the very difficult to remove sulfate ions and lowering of
sulfate-based salinity by 90 to 99% or more,
4) Lowering of water total dissolved solids,
5) Achieving the above separations simultaneously and quickly, thereby
achieving a process with a relatively very small size footprint for high flow
rate systems,
6) Significantly, by means of this invention, highly toxic contaminants such
as
Pb, As, Se, Hg, Cd, and the like; and many radioisotopes/radioactive
contaminants such as Tc, I, and the like can be effectively removed from the
water,
7) Purification even of waters contaminated with extremely problematic,
complex mixtures of contamination ions in waters containing slowly-reacting
43
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
ions, colloids, highly-toxic metal ions, and up to thousands of mg/L (ppm) of
dissolved salts.
A particularly problematic example is that of mining, especially that of
coal mining, where the mining activity has exposed gangue minerals left-in
the mine and mine tailings to erosion by air, water, and microbial action.
Sulfidic minerals, such as pyrite, FeS2, are commonly found in many
geological strata, and especially in reducing ores such as coals and metal
sulfide ore bodies. As described thoroughly in the literature review spanning
many decades, coal, metal and other mining operations, and natural
m weathering fissures have allowed water, air and microbial access to these
reducing substances. These conditions promote the oxidation of the sulfidic
minerals to water soluble metal sulfates, especially ferrous sulfate solution,
as
shown by the series of chemical equations below. In the specific case of iron,
a complex series of reactions can occur (3. Skousen, et al, June 1, 1998,
.. "Acid Drainage Technology Initiative (ADTI)," Published by The National
Mine
Land Reclamation Center located at West Virginia University in Morgantown,
West Virginia, Handbook of Technologies for Avoidance and Remediation of
Acid Mine Drainage) with the net total dissolved solids level and acidity
increasing result as follows (aided by microbial action):
2 FeS2 + 7 02 + 2 H20 --. 2 Fe2+ + 4 50.42- + 4 H+ (10)
4 Fe2+ + 02 + 4 H+ -. 4 Fe3+ + 2 H20 (11)
4 Fe 3+ + 12 H20 --. 4 Fe(OH)3* + 12 H+ (12)
FeS2 + 14 Fe3+ + 8 H20 --. 15 Fe2+ + 2 5042- + 16 H+ (13)
4 FeS2 + 15 02 + 14 H20 --> 4 Fe(OH)3* + 8 H2SO4 (14)
* solid at pH > 2.5
These reactions produce an acidic ground water of primarily ferrous
sulfate, the main soluble metal ion product, containing very high levels
(often
above 1000 mg/L) of total dissolved solids, being largely a contribution of
acid
attack on dolomite and limestone carbonate-based minerals (Reaction 15 and
16 respectively) which contribute Mg, Mn and Ca, which also report to the
water in sulfate form. Though bicarbonate ion can also be present in some
44
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
alkaline acid mine drainage waters, waters of pH less than about 6 will have
converted most of the carbonate of these minerals into gaseous CO2 and so
little alkalinity will is found.
2H2SO4 + MgCa(CO3)2 --> 2H20 + 2CO2(9) + Ca2+ + Mg2+ + 2504 (15)
From dolomite
Eq. 14
H2504+ CaCO3 4 H20 + CO2(g) + Ca2+ + SO4= (16)
From limestone
Eq. 14
Hence, due to Reactions 12 and 14, acid mine drainage, acid rock
drainage, natural gas well brine, and the like, waters will tend to have far
more dissolved sulfate ion than dissolved iron, the pH will be higher than
Reactions 10 to 14 would predict alone, and would be low in ferric ion
content. Hence, for the maximum benefit, the technology of this invention is
best applied to the acid mine drainage water drawn directly from the mine
pool and not from the discharged surface waters in that much more of the
metal ions can be recovered instead of lost to Fe0OH precipitation in the
affected natural stream or other water body.
These drainages and associated ground and surface waters contain
one or, more often, many of the toxic or highly concentrated contaminants
listed in Tables 2A, 2B, and 2C which provide an analysis of a representative
samples of acid mine drainage.
Conventional technologies have been found to be unsuitable for
purifying such waters as acid mine drainage, many mineral processing waters,
gas well brines, and the like. Typical concentrations of the contaminants of
concern in such waters are several fold above the 1000 mg/L removable by
such prior art technologies and the ferrous ion rapidly air oxidizes to Fe0OH
precipitate rapidly plug alternative technologies. The flow rates of such
ground waters, especially as they seep to the surface and contaminate
ground water for wells, streams and rivers far exceed the practical treatment
maximum of these alternatives.
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
= Industrial processing of metals (surface finishing). These plating and
painting shop rinse waters contain one or more of the toxic or highly
concentrated contaminants listed in Tables 2A, 2B, and 2C and also toxics
such as hexavalent chrome, cyanide ion, lead, cadmium, nickel, copper, iron
and others.
= Discharges of agricultural water from concentrated animal feeding
operations, CAFOS, and the like. These drainages and associated ground and
surface waters contain one or more of the toxic or highly concentrated
contaminants listed in Tables 2A, 2B, and 2C, and more particularly also
contain problematic macronutrients, especially phosphorus (P) and nitrogen
(N) containing organic and inorganic compounds. P and N nutrients are the
bane of agricultural runoff waters because they stimulate plant algal, and
then bacterial, growth that far exceed natural waters' ability to handle it,
resulting in deoxygenated zones ("anoxic" zones) in natural bodies of waters
extending from the farm, such as streams and creeks, leading to ponds, lakes
and rivers, and then to large lakes, like Lake Erie, gulfs, like the Gulf of
Mexico, and bays, like Pamlico Sound (all in the USA). These dissolved
oxygen "dead zones" are lethal to aquatic fish and thereby render these
streams and agricultural practices problematic to the public recreational,
fishing industry, and government regulatory bodies.
Most phosphorus and nitrogen containing compounds in fresh manure
and urine animal waste, or from animal feed decomposition processes taking
place in storage bins, occur as oxidized phosphate in the case of P but
reduced amines in the case of N, the later some inorganic, primarily
ammonium ion, and the rest as well known biochemical organo amine
compounds. It is also well known that the P component in agricultural runoff
water is by far the most impactful micronutrient on algal blooms. Since the
present invention readily extracts anions, like phosphates that are present in
these problem waters, a substantial improvement in water quality emanating
from farms can be made using the invention. Likewise, after operating the
residual organic and ammonical nitrogen components oxidatively using
established physical chemical and/or biological operations to convert the
46
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
nitrogen to the nitrate anion, then the nitrogen component could also be so
removed from the water and concentrated for fertilizer use, for example as
needed by international grain grower farms. In this manner, agricultural
waters can be purified to enable their problem-free discharge to surface
s waters without the negative impacts described above due to the payload of
nutrients. Most preferred is to first filter out any suspended solids by
conventional filtration prior to treatment according to the present invention.
Method (Operation Of Process)
Acid mine drainage/acid rock drainage feed water is used to illustrate
application of the preferred operation of the invention. Coal mine acid mine
drainage water (Tables 2A, 2B, and 2C) is typically most contaminated with
the metal ions: ("M" Series) Fe(III), Al, Cu; ("N" Series) Fe(II), Ni, Co, Zn,
Mn, Ca and Mg and highly contaminated in sulfate anion. Sulfate ion is a
notoriously difficult anion to remove from water, and especially in a
practical
manner for the cases of unmet need involving high to very high flow rates
(e.g. 10 to 10,000 gal/min). Typical acid mine drainage water contamination
and flow rate levels vary broadly by source, season, and physical
characteristics of the aquifer involved. This invention enables the removal of
most or all of these contaminants to levels low enough to allow widespread
use of the water no matter the variations in inlet contamination and flow rate
levels. Contaminant levels can usually be reduced to below maximum
permissible dischargeable levels in one treatment, and even routinely to
levels
allowed by primary and secondary government drinking water standards.
Therefore the product water is safe for aquatic life promoting tourism
involving such river and stream systems. Even more, such purity levels would
enable the product water of the invention to be used for potable water, feed
for potable water production plants, farm water use, and/or for industrial
process water. As described below, the ability for this high level of
purification is provided by the invention via a novel combination of new
process chemistry, newly designed simultaneous floc/liquid/liquid contactor
devices, and the newly discovered process operations needed to deploy these
47
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
,
technologies. Referring now to Figures 1A and 1B, a detailed description of
this integrated invention follows.
As used herein the term ionic ions species includes one or more of an
anionic and/or a cationic ions.
A broadly applicable and detailed version of the invention is given in
Figures 1A and 1B. This water purification process provides new and useful
process chemistry, water purification devices, and methods. The major
features of the invention, the operation of which is detailed in the example
below, include a novel flocculation-liquid-liquid extraction device that
io possesses a unique design for floc and slurry handling hardware to
enable
metal hydroxide/sulfate colloids and flocs to be handled in modified liquid-
liquid contacting devices. Such flocs and slurries rapidly shutdown
conventional and other prior art liquid-liquid extraction apparati. In one
embodiment, this unique hardware consists of a liquid-liquid contacting device
fitted with a extractant phase flow control consisting of gradually narrowing
channeling gates feeding it to a smooth, preferably about 30 degrees sloped-
surface, that feeds it to a smooth and broadly rounded over-flow type weir
called a floc weir fitted with a sloped discharge ramp of most preferably
about
30 degrees from the vertical. These features enable the colloids but
especially flocs and slurries produced by the unique process chemistry of the
invention to be separated from the water phase being purified in the process.
The unique separation process chemistry of the invention is normal very
difficult to process but is required to achieve very short water residence
times
in the equipment which in turn provide the enormous advantage of purifying
very large volumes (10 to at least 10,000 gal/min) of water practically and at
much lower cost than conventional technologies.
Referring to Figures 1A and 1B the detailed description of the invention
is as follows. Acid mine drainage water 1110, containing anionic and cationic
components, is fed by gravity or by pump 1120 through valve 1125 and line
1130 to one or more floc/slurry liquid- liquid extractors 1210 fitted with
specially designed floc/slurry handling decanters as described elsewhere in
this application. The level of contamination and the degree of water
48
CA 02911119 2015-11-03
40 WO 2008/100610
PCT/US2008/002092
purification desired determines the number of such extractors deployed.
Normally the selection of the number of stages and flow rate ratios used is
made by the level of purity desired for the product water, especially the
residual sulfate ion and toxic metal residuals needed. Most preferably the
arrangement of such extractors is counter-current, but can be preferably
cross-current, or least preferably co-current. If more than one contactor is
provided, they are most preferably arranged in counter-current liquid-liquid
extraction configuration. This water is contacted only for a short period of
time in each extractor, 30 seconds to 30 minutes are effective, however,
preferably only 30 to 200 seconds, and most preferably about 45-90 seconds,
and still me preferably 60 seconds, and still more preferably 60 seconds, with
extractant phase (defined elsewhere in this application), supplied from tank
1010 via pump 1020 and valves 1030, 1032 and 1034.
Water Purification Using Only One Extraction Operation
In the simplest case, where only mostly Fe(III), (including Fe(II)
converted to Fe(III) during processing), Al, Cu, CM" Series of metal ions) and
a substantial amount of sulfate ion needs to be removed from the water to
accomplish the purification objective, then only the Low pH flotation liquid-
liquid extractor process arrangement is used. In this case at least one
flotation liquid-liquid extraction extractor set 1210, containing at least one
mixing stage forming at least one water immiscible heterogeneous fluid
consisting of liquid (at least two phases consisting of an aqueous and water
immiscible extractant phase (E-phase), and also a floc and/or colloidal
emulsion, and/or optionally a solid/liquid slurry. Also included is at least
one
settling compartment, optionally, but not preferably, fitted with "picket
fence" fluid flow disrupter gates, and where the settler compartment
necessarily contains a flow constrictor positioned to guide the top phase
(organic emulsion containing colloidal floc and/or slurry) towards the
specially
designed floc/extractant phase overflow weir at the back of the settler. In
this manner extraction of at least a portion of the above contaminants from
49
CA 02911119 2015-11-03
a WO 2008/100610
PCT/US2008/002092
the acid mine drainage (or other) feed water is accomplished using the
extraction process chemistry described elsewhere in this application.
The metal ion and sulfate ion "loaded" extractant phase from 1210
exits the settler as a water insoluble floc and/or slurry commingled with
liquid
extractant phase optionally, but most preferably, to a decanter. The
construction of the decanter (for Figure 1A is to be presumed to be a
component of F-LLX Extractor 1210) is similar to the settler and includes the
floc handling overflow weirs described above. However, the most preferred
operation of the Extraction 1210 settler and associated decanter in that the
m E/A interface of the settler is set high in the extractor settler to
promote
exiting of the E-phase and its contents as quickly as possible to the
decanter,
and low in the decanter to facilitate releasing the bulk of the water from the
extractor settler as fast as possible but releasing the extract with floc
slowly
from the decanter to maximize its dewatering. This short residence time is
valuable since the acid mine drainage water flow rate is by far the fastest
flowing fluid in the system (at least 5 to 10 times any of the other flows)
and
therefore dictates the size of the hardware need to make the purification.
The E-phase with floc is dewatered maximally in the decanter prior to
metal product production. This further dewatering is achieved by setting the
zo E/A interface at a medium position in the decanter, normally in the 1/3
to 2/3
range level of the total fluid depth of the decanter. This interface
positioning
allows a very clean/sharp separation of the water from the floc material in
the
decanter. It also allows the E-phase to thicken as it releases water of
entrainment, chemically formed water from the dehydration of metal
hydroxide colloids, chemical displacement of hydration water by the modifier
(normally oil soluble isodecanol or other oil soluble yet polar component of
the E-phase, such as esters and/or alkyl phenols). Critically, the fluid
dynamic
design of the decanter internals provide the necessary promotion of
continuous thickened E-phase floc fluid flow in the decanter which, if such
devices were not present, would result in settler and decanter plugging by
heavy precipitates causing stoppage of flow. However, with such newly-
discovered devices, the E-phase emulsion thickens, but does not collect at the
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
interface (known as "CRUD" in the conventional technology and prior art,
resulting in the requirement for maintenance and even shutdown/cleanout),
but instead flows smoothly and continuously to the E-phase exit chamber
1040, which can be a line, a chute, or the like, as described elsewhere in
this
s application, thereby transferring it to the metal ion stripper 1220
operation.
The acid stripper operation 1220 consists of one or more, most
preferably counter-currently arranged liquid-liquid contactors, which can be
of
conventional design, but which most preferably is designed capable of
accepting flow of floc/slurry impregnated E-phase to the mixer compartment
io via 1040. In the case where more than one stripper contactor exists in
counter-current arrangement, this modification is only needed for the first
mixing compartment as the floc or slurry is rapidly dissolved in the first
strip
mixer operation.
In the strip mixer operation, the floc/slurry loaded E-phase is contacted
15 .. with aqueous acid solution 1215. This acid is delivered to the stripper
contactor via pump 1217 and valve 1219. Most preferably, the strip acid is
continuously internally recycled within each striper liquid-liquid contactor
used
in order to achieve both efficient use of the acid, by consuming it as
completely as possible, and the maximization metal ion concentration in the
20 metal sulfate product solution. The greater the metal ion concentration
of the
aqueous strip solutions the more useful and valuable is the metal sulfate
product and the more cost effectively it can be processed into items of
commerce. With the invention, the extracted metals (and later sulfate, see
below) can be concentrated many factors, for example from 2X to 200,000
25 times, and often to saturation points for the metal sulfate, carbonate
or
hydroxide solids produced (see below). It will be appreciated that the lower
the concentration of solute in the acid mine drainage feed water, the greater
the concentration factors that are theoretically possible. For example, 2000
ppm sulfate ion present in feed water 1110 can be concentrated by the
30 invention to a sulfate solution concentrate in operation 1370 of 200,000
ppm,
representing a concentration factor of 100X. However, 200 ppm sulfate ion
present in feed water 1110 can also be concentrated by the invention to a
51
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
sulfate solution concentrate of 200,000 ppm, representing a concentration
factor of 1000X. In this manner, valuable metal ions, for example Co or Ni,
that are present at sub-economic concentrations, for example 0.1-1 ppm, can
be concentrated to 1000 ppm in operation 1220, a 1,000-10,000 X
concentration factor, making these metals now economically and practically
available for commercial use. The invention achieves these attractively high
concentration factors by using a combination of sharp separation process
chemistry both for extraction and stripping, low E/A ratios in the extraction
stages, and then high E/A ratios with aqueous internal recycle in the strip
to stages, in the stripper stages 1220 and 1250. The amount of recycle
acidic
metal sulfate is maximized and controlled by valve 1240.
The aqueous acid, now carrying the metal extracted from 1210 and
representing metal sulfate concentrate exits the metal ion stripper 1220 via
line 1230. If the metal concentrate is concentrated sufficiently for
harvesting
is and/or there is a need to drain aqueous phase from stripper 1220 to
prevent
excessive aqueous phase volume accumulation in the stripper settler
compartment, then this product metal sulfate aqueous concentrate is
harvested via valve 3-way valve 1240 to allow flow of this metal sulfate
concentrate to optional oil/water separator 1270. Any recovered extractant
zo phase from oil/water separator 1270 is returned to the extractant phase
exiting the metal ion stripper circuit 1060. In this manner, especially
preferred to be a counter-current multi-stage design, the E-phase is rendered
devoid of most of the metal ions extracted above in the extraction operation
and now proceeds to the sulfate stripping operation via line 1060. The metal
25 sulfate concentrate exits optional o/w separator 1270 via line 1280 and
proceeds to optional solid/liquid separator 1285, for example a crystallizer,
in-
line filter, or other solids/liquid separator, the liquid proceeds to M metal
sulfate concentrate through line 1290 to metal sulfate product collection
vessel 1292. The optionally crystallized product or filter cake is collected
via
30 line 1287 to receiving vessel 1289. The extractor and stripper can be
one
contactor each or more than one. If more than one, then they are most
preferably configured counter-current or cross current. The concept of
52
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
counter-current, cross-current and co-current are already well known to those
skilled in the art of industrial scale liquid-liquid extraction. For the
invention,
one or two stages of flotation liquid-liquid extraction each, with decanter is
most preferred when the objective is to remove only ferric ion, aluminum,
and/or copper with two, or preferably three, stages of metal ion stripping
operated with internal recycle in the mixer E/A range of 1/20 to 20/1, and
with the aqueous acid feed fed most preferably counter-current or cross-
current to the strippers and while the aqueous phase from the third metal
sulfate strip (S3M) stage is being sent to the second metal sulfate strip
(S2M)
to stage, then being sent to the first strip stage (S1M) as aqueous phase
of
metal ion sulfate concentrate from the first stage (S1M) is harvested.
Decanters are not needed for the strip stage so long as the settler volumes
are sufficient as is normally the case. Most preferred is that at least 15-30
min
of metal sulfate stripper settler time is supplied. Although a third phase can
.. form in the S2M stripper settler, this can be alleviated by altering the
composition of the extractant phase or most preferably by lowering the
concentration of the strip acid to about 25%, though 2% - 30% acid
concentrations are effective.
After metal sulfate stripping operation, the extractant phase 1060, now
zo .. loaded with bisulfate ion and devoid of at least a portion of the
transition
metal and light metal ion content, and preferably devoid of > 90% of the
transition metal and light metal ion content, and more preferably devoid of >
99% of the transition metal and light metal ion content, and most preferably
devoid of > 99.9% of the transition and light metal ion contents, flows
preferably by gravity through line 1080 to the sulfate stripper operation 1370
where it is stripped of its anion content, sulfate ion in this example of acid
mine drainage water purification, using one, or preferably 4 to 6 counter
current, internally aqueous recycled, stages. Sulfate stripping is
accomplished
in operation 1370 by contacting it counter-currently with an aqueous solution
of carbonate ion, hydroxide ion, a combination of the two, and/or bicarbonate
solution, carbonate ion solution, or a combination of the two. Suitable
cations
for these anions are most preferably sodium, potassium or ammonium ions,
53
CA 02911119 2015-11-03
,
WO 2008/100610
PCT/US2008/002092
or less preferably, lithium ion, or any combination of these. Sodium cations
and/or potassium solutions are the most preferred, i.e. Na2CO3 solution,
and/or K2CO3, with or without KOH or NaOH. Although one mixer-settler is
functional, preferably three to six mixer-settlers arranged in counter-current
flow is more preferred, and five is most preferred. The criteria for the
number of stages to use are the amount of sulfate ion in the feed water, the
E/A ration in the sulfate strip circuit, and the target low level of residual
sulfate ion sought in the finished water. Any residual sulfate ion left on the
E-
phase after sulfate ion stripping operation is carried back into the
extraction
operation with the acid mine drainage feed water and hence may reduce the
sulfate ion removal efficiency of the extraction operation and therefore the
residual sulfate ion concentration left in the purified water product.
For illustration, using the sodium cation case for the carbonate
aqueous feed 1310 to the sulfate ion stripper 1370, and the case of no NaOH
feed 1360, fed to the sulfate stripping operation 1370 via pumps 1320 and
1350 via valves 1325 and 1345, the sulfate ion is removed from the
extractant phase into the aqueous phase where it forms a sodium sulfate salt
concentrate at a ratio depending on what was fed via lines 1330 and 1340 via
pumps 1320 and 1350 from reagent feed tanks 1310 and/or 1360. During
this operation, the extractant phase becomes loaded with carbonate ion,
bicarbonate ion, and/or hydroxide ion, depending on the E/A flow ratio in the
strip circuit and the setting of valves 1325 and 1345 and the speed of pumps
1320 and 1350. For removing the above listed acid mine drainage metals
from the extractant phase, other than Mn, Ca, or Mg, preferably only
carbonate ion, is used to strip the sulfate ion from the E-phase 1060 and/or
1080 (see below), and is fed from tank 1310 with pump 1320 via valve 1325
and line 1330.
Note, that with bisulfate ion, HSO4- loaded onto the E-phase flowing
from the M metal sulfate stripper as described above, the first stage of
sulfate
stripping involves converting extracted bisulfate ion into extracted sulfate
dianion as follows,
54
CA 02911119 2015-11-03
WO 2008/100610 PCT/US2008/002092
2(R4NHSO4) + Na2CO3 4 (R4N)2804 + CO2T + H20 + Na2SO4 (17)
Where, the second stage of sulfate stripping, and any additional stages
supplied, accomplishes essentially complete sulfate ion removal by ion
exchange, i.e.
(R4N)2SO4 + Na2CO3 (FZ4N)2CO3 + Na2SO4 (18)
Where maintaining low E/A ratios in the stripper mixers enables the
build up of very high sodium sulfate concentrations (e.g., 40,000 to 650,000
ppm as SO4= where 150,000-250,000 ppm is preferred for the case of a
Na2SO4 product, at 10,000 ¨ 100,000 ppm, and, preferably 40,000 - 60,000
ppm, for the case of a K2SO4 product. Higher concentrations are possible if
preparations are made to harvest the resultant slurries of crystals formed.).
Complete (efficient) usage of the carbonate is achieved. Notice that the
humid 1371 CO2(g) product produced above can optionally be collected from
the headspace of the sulfate strippers (especially from the first stage of
stripping, S1-SO4, in a multi-stage operation). This CO2 gas is a weak acid
with natural buffering tendency for the pH range of about 6-9 and so, if
zo needed, can be used to bring the pH of the product water from pH > 9
into
this pH range (see below) before discharging it to natural streams, used in
agricultural operations, used as feed to potable water plants, used in
industrial operations, and the like. As will be shown below, high (> 9) pH
product water is produced only when > 90% or the Mn, Ca and/or Mg are to
be removed by the invention where pH values of the treated water can be
above 9, and as high as 12.
Once harvested, the high concentration of the sodium (or potassium)
sulfate product stream exits line 1384 to optional 0/W separator 1387
through optional valve 1380. Valve 1380's main purpose is to prevent
premature harvest (aqueous flow) to help insure full conversion of bisulfate
to
sulfate dianion in S1-504 stage. Any E-phase captured by the 0/W is
returned to stage S2-504 in the usual manner (0/W E-phase flows are not
CA 02911119 2015-11-03
WO 2008/100610 PC
T/US2008/002092
shown in Figure 2 to minimize clutter diagram, since their role and function
are well known in the art, and because they normally carry very little flow).
The extractant phase return to the extractors/strippers is always to the next
stage downstream of the stage where it was generated.
Exiting the 0/W separator the sulfate concentrate flows to an optional
solid/liquid separator 1390 via line 1389 if it is desired to remove sodium
sulfate crystals, potassium sulfate, or other solids such as metal oxides
and/or
metal from the concentrate. Such crystals are collected in product vessel
1392. The mother liquor then can be collected as liquid concentrate in tank
lo 1398. Alternatively, both products could be collected as slurry in tank
1398
by by-passing the solids/liquid separator 1390, thereby obviating the need for
line 1394 and vessel 1392. After exiting the solid/liquid separator via line
1393, the sulfate concentrate flows through an optional trace E-phase
sorbent, such as granular activated carbon (GAC) 1391 and/or other
hydrophobic sorbent materials, then exits via line 1396 and is collected in
tank 1398.
As the sulfate concentrate has a very high ionic strength, little if any E-
phase contamination of it should occur. And then, any entrained E-phase
should have been captured by the 0/W separator 1387. Hence the use of
(GAC) 1391 is not expected to be needed in most instances but if used, then
GAC use rate is minor and is used to remove any E-phase odor from the
sulfate product.
The pH < 5 raffinate exits extractor circuit 1210 via line 1420. Before
releasing this water, it is most preferred to pass the water through 0/W
separator 1460 via valve 1430 and line 1450. The 0/W separator effluent
1470 can optionally be filtered and deodorized via solid/liquid separator 1480
and GAC 1495 to produce clear, colorless and odorless water in line 1496 that
is used immediately or collected in a storage/surge vessel 1494. The minor
amount of particulates collected 1492, normally of a rust-like character, via
line 1491 are nontoxic and can be discarded.
56
CA 02911119 2015-11-03
,
WO 2008/100610 PCT/US2008/002092
Water Purification Using Two Extraction Ooerations
In certain cases, depending on the requirement targets for the purified
water, it may be desirable to remove trace metal ion contaminants of the
mostly divalent metal ions, for example Ni, Co, Zn, Fe(II) as well as
(the "N" series of metal ions of Figure 1A) and/or to achieve even greater
removal of sulfate ions. Additionally, it may be desired to separate these
metals from those collected in the pH < 5 extraction operation described
above. In these cases two extraction operations, each with one or more
liquid-liquid contactors, are recommended and provided. This second
operation is now described with the two variants mentioned.
The first operation is as given above and results in an extractor mixer
pH and raffinate pH of about 5. Adding a second extractor circuit 2030,
which receives ¨ pH 5 water from the first contactor circuit 1210 via the
combined settler and decanter aqueous overflow line 1440. The E-phase is
fed to the second extraction operation from the common E-phase surge tank
,
via line 1025 and pump 1020 and opening valve 1032. To operate cross-
current, valve 1030 is open three way such than E-phase simultaneously flows
to both extraction operations 1210 and 2030. Alternatively, 1030 can be
closed to 1210 and open to 2030, so that now first extraction circuit 1210
receives extractant phase from second extraction circuit 2030, i.e., a counter-
current arrangement.
If ferrous ion is included in the acid mine drainage water feed 1110
then it is normally present in large amounts relative to ferric ion. Hence
extraction operation 2030 can again form a large amount of floc of divalent
metal ions and so this contactor circuit is also of the specially designed
flotation liquid-liquid extraction type of the invention, described in detail
above, and is accompanied by a flotation liquid-liquid extraction decanter,
also of the invention and previously described.
The operation of the second unit is as follows. Depending on the E/A
ratio, number of contactors per circuit, the concentration of extractant in
the
extractant, and/or the number of equivalents of the above metals present,
the final pH of the extraction can be about 7.5 to 9. This condition
efficiently
57
CA 02911119 2015-11-03
WO 2008/100610
PCT/11S2008/002092
removes divalent metals which can be only partially removed by the first pH
-5 extraction. The attainment of pH 9 helps Mn removal to about 90% and
this higher pH can be promoted by including some hydroxide ion on the
extractant that can be provided from supply tank 1360 via pump 1350 and
valve 1345 through line 1340. It is one important aspect of this invention
that basicity is brought into the extraction process without the
accompaniment of water soluble cations. This accomplishment is made
possible by using the water insoluble cationic quaternary ammonium ion salt
of basic anions. This feature enables the substantial decrease of the total
dissolved solids of the product water relative to the feed water.
The pH 7.5-9 raffinate, now clear of substantial contamination can
represent very desirable product water. Before releasing this water, it is
optionally, but most desirable, to pass the water through 0/W separator 2090
via optional valve 2070 and line 2080. (Valve 2070 is only needed when
additional extraction stages are added as described below). The 0/W
separator effluent 2110 can optionally be filtered and deodorized via
solid/liquid separator 2120 and GAC 2125 to produce the clear, colorless and
odorless product water in line 2140 that is used immediately or collected in a
storage/surge vessel 2150. The minor amount of particulates collected 2130
via line 2115 are nontoxic and can be discarded. Due to the scale of some of
the acid mine drainage water streams, the filters used could be sand filters,
clarifiers, simple in-line filters such as plate and frame, drum or belt
filters,
centrifuges, and the like. The GAC is again for odor control of clear water
product and so is optional and represents a small use rate.
The metal and sulfate ion loaded extractant phase can be process
either of two ways according to the invention (for convenience the two
options will be referred to as options AA and AB). These can be operated
simultaneously, alternated with acid mine drainage feed content, economic
conditions for the products produced, and so on.
In option AA, the loaded extractant is merely sent forward to the first
extraction phase via line 2040 and where valve 2042 is open to flow to the pH
5 Extraction operation but closed to line 2044. In this mode, valve 1030 is
58
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
optionally closed so that the entire extractant phase flow is through valve
1032 to the pH 7.5-9 Extraction operation. In this manner all the metals
extracted report to the metal sulfate extractor forming a combined "M+N"-
Sulfate concentrate 1292 in the identical fashion described above for the M-
s Sulfate product. After stripping of the metals, the sulfate is stripped
in a
fashion also identical to that described above previously.
Option AB differs in that it provides separation of M-sulfate and N-
sulfate products using cross-flow contacting of E-phase with the acid mine
drainage water and stripping the two loaded phases separately forming two
concentrate products, M-SO4 and N-SO4. In this case pH 5 Extraction
operation is performed exactly as described above including receiving E-phase
feed via valve 1030 and producing M-SO4 product 1292. However, the N-
loaded E-phase formed in 2030 is sent to a separate acid stripper 4000 via
line 2044 by diverting the flow from 2030 using valve 2042. In this case a N-
Sulfate concentrate is produced in a fashion identical to M-Sulfate and using
equivalent process hardware, including internally recycle aqueous via line
4020 and with the stripped extractant phase flowing forward to sulfate
stripping 1370 via lines 1070 and 1080. Hence the N-Sulfate concentrate,
when harvested, is sent via line 4010 and valve 4030 (which again can just be
an overflow weir), through an optional 0/W separator 4040 to an optional
solid/ liquid crystallizer 4060 via line 4050 if solid product crystals are to
be
separated from liquid product concentration (not preferred). The solid
crystals can be collected in vessel 4070 via line 4080 and the liquid
concentrate in tank 4090 via line 4095. Most preferred is just to collect one
product slurry into tank 4090. Deodorizing is again optional (and not
normally needed or preferred), for example by GAC treatment.
Whether option AA or AB is preferred can depend upon the level of
Fe(II) in the acid mine drainage water relative to Fe(III). If a high level of
Fe(II) exists (this is often the case when acid mine drainage feed water is
derived directly from wells or abandon coal mine shafts), then this Fe(II)
will
be extracted in pH 7.5-9 Extraction operation and therefore the more valuable
but dilute "N" metals, i.e. Ni, Co, Zn and Mn, will be collected with the
large
59
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
volume of Fe(II)-sulfate concentrate produced (if air oxidation of Fe(II)-
Fe(III) is prevented by limiting air access to the extractor mixers, this is
easily
accomplished by simple tank covers and by not sucking air into the feed lines
to the mixers). Hence if there is very little Fe(III), Al and Cu present ("M"
metals) then there is no motivation for operating two separate stripper
circuits and collecting all the metal sulfates in one product as a
"concentrate"
that can be further refined into saleable products in a side and much smaller
operation is preferred. Hence the acid mine drainage feed water composition
determines the most preferred mode of operation.
It is noted that in the above pH 7.5-9 operation, the Mn is only about
90% removed. Hence, depending on the fate of the product water (discharge
or feed to potable water production plant, etc.) it may be desirable to add a
third water purification operation (see below).
Water Purification Using Three Extraction Operations
The need can arise for a third purification extraction operation in some
situations where the water is to be used rather than discharged. In acid mine
drainage this arises where the intended use for the purified water is for feed
to a potable water production plant, and industrial operation, and the like,
where hardness metal ions, still lower TDS, lower alkalinity and/or less Mn
contamination levels reduction is desired.
For example, to reduce Mn levels to below secondary drinking water
standards of 0.05 ppm for the case where the acid mine drainage feed water
level is sufficiently high that this Mn residual level is not attained in the
raffinate from the first two extraction operations 1210 and 2030 despite
removing 90% of it. In this case, the pH of the water needs to be adjusted to
higher pH to convert the Mn to MnCa(CO3)2 particulate that can be filtered
out. As the acid mine drainage feed water is already far excessively
contaminated in total dissolved solids, e.g. several to ten times the level
permissible for drinking water. It is needed to make this pH increase
adjustment without the addition or formation of additional salts in the water.
The invention provides this unexpected capability as will be described below.
CA 02911119 2015-11-03
.
WO 2008/100610
PCT/US2008/002092
In addition, if the acid mine drainage feed water was initially very high in
sulfate ion concentration, there may be a need to remove the sulfate ion
concentration to still lower levels to enable use, rather than discharge, of
the
water.
Due to additional floating floc formation involving the Mn, Ca and/or the
residual sulfate ion, the third extraction step is also most preferably
performed as another flotation liquid-liquid extraction operation (see above
for description of the first two flotation liquid-liquid extraction
operations).
The preferred configuration for these additional two steps is the flotation
liquid-liquid extraction operation but without a decanter. Decanters are no
longer needed since floc formation is substantially reduced in the subsequent
contacts relative to the first two extractions. However, instead, particulates
form and the low ionic strength and higher pH of the water product after a
third extraction operation still higher pH results in at least a portion of
the
contacted fluid exiting the mixer compartment of 3020 via line 3050 and valve
3060 to be a emulsion that requires additional processing to separate (Figure
1A and Figure 1B)). This unstable emulsion is an ionic colloidal complex
between the MnCa(CO3)2 solid particulates and the Aliquat-SO4= species.
Hence a solid/liquid (S/L) and liquid/liquid separation is needed. Such
zo separator can be a filter, a semi-continuous centrifuge, hydrocyclone or
other
solid/liquid (S/L) separating device. The extractant phase formulation can
also be adjusted to encourage separation of these emulsion components for
example by increasing modifier concentration change diluent, blend diluents,
and the like.
Freshly prepared extractant phase can also produce a "milk" colored
aqueous phase that is believed to arise from hydrophobic tertiary amine
complexes that form from E-phase manufacturer impurities at the higher pH
of the third extraction due to deprotonation of the amine. This milk does not
contain significant amounts of Mn and can be discarded. The extractant can
also be purified of tertiary amine impurities prior to using the extractant in
the
first place by acid washing, washing with copper sulfate solution, and the
like.
How these process features are performed is described below.
61
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
This third extraction operation is accomplished by sending the pretreated
water from the second puriflcation operation described above (using either
options AA and/or AB) via valve 2070 to line 2060 to flotation liquid-liquid
extraction (or conventional LLX) contactor 3020.
The pH of the third extraction is in the 8.5 to 10.5 range. This higher pH is
accomplished by including hydroxide ion in the extractant regeneration
operation of sulfate stripping. Most preferably, NaOH is used since it forms
the far more soluble Na2SO4 product 1398, but KOH can also be used if more
dilute sulfate product is acceptable. The hydroxide is fed to the sulfate
lo stripper from tank 1360 via pump 1350 and valve 1345 through line 1340.
At the higher pH, the concentration of carbonate ion becomes
significant causing the particulates of MnCO3 to form; with any Ca present
forming CaCO3. Mg is not removed at these conditions requiring higher pH
(see below). Particulate formation is desirable as it allows the removal of
these Mn and Ca contaminants with a corresponding decrease in total
dissolved solids since the OH- can be introduced from via the cationic E-
phase. The overall reaction is,
Mn2+ + 2HCO3" + SO4= + Ca2' + 2(R4N1.01-)E-phase
MnCO3 particulate + CaCO3 particulate + ((R4N)2SO4)E-phase + purified waterpH
8.3-10.5
This product mixture forms an extractant with floc and an aqueous phase
low in Mn, Ca and SO4= content that exits contactor 3020 via line 3050 and
valve mixture exits the 0/W separator 3040 via line 3065 and enters the
solid/liquid separator 3070. In the solid/liquid separator 3070, the Mn(C0)3
and Ca(C0)3 solids 3090 are separated from the mixture via line 3080. The
extractant phase is sent via line 3075 to the second extraction operation 2030
for additional stripping and regeneration. Although most of the E-phase
separates from most of the aqueous phase, the low ionic strength of the
product mixture, and the high ionic character of the quat:sulfate ion cluster
results in some formation of a micro emulsion milky white product water.
Analysis of this micro emulsion shows it to be low in Mn and SO4'. The micro
emulsion is believed to be caused of surface active impurities in the Aliquat
62
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
134, and these are known to wash out from the system over time, and so is a
non-issue.
The extractant phase floc, a brown emulsion, contains the extracted Mn, Ca
and sulfate ion. This floc is dewatered and thickened in the decanter as was
done in the first two extraction operations. The E-phase floc then could be
stripped of metals using acid as before, filtered, centrifuged, or other
solid/liquid separation 3070 to gather the Mn and Ca in particulate form 3090
via line 3080. The extractant 3075 is then sent to sulfate stripping directly
(preferred) or to the second extraction operation, or to a fourth extraction
io operation (see below). The stability of this emulsion could also be
reduced
using a weaker (particulate) or stronger ion pair solvating E-phase
formulation.
The pH 8.5 ¨ 10.5 raffinate exits the solid/liquid separator 3070 via line
3075 and valve 3100. The pH of the purified water can be reduced to the pH
6 - 9 range by sparging inline 3105 with the CO2 gas 1371 via line 1372 and
valve 6000. In addition, clarified raffinate 3110 can be sent to contractor
5000 for Mg removal. The pH 6-9 raffinate exits valve 3100 via line 3105 and
enters GAC 3120 for deodorization. The deodorized and purified water exists
GAC 3120 via line 3125 is collected in a storage vessel 3030.
Water Purification Using Four Extraction Operations
The purified water from the above-described four extraction stages is
fairly pure. However, if Mg levels are significant then dissolved Mg sulfate
and carbonate salts still would be present in the water. In the case of acid
mine drainage this can represent hardness levels in the range of about 400 ¨
900 ppm, too high for certain end use applications. For example for the case
of add mine drainage water with 140 ppm and 1600 ppm SO4', 130 ppm Mg
and 135 ppm sulfate can still exist in the water giving rise to high total
dissolved solids residuals.
Although Mg is not removed efficiently in any of the above-described
flotation liquid-liquid extraction operations, it was discovered that Mg can
be
removed without employing conventional water softening by using a flotation
63
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
liquid-liquid extraction operation 5000 at still higher pH where the hydroxide
ion is again introduced by the E-phase via valve 1034. It was discovered that
in a flotation liquid-liquid extraction operation, hydroxide ion can be
effectively brought in with the extractant phase (added at the 1370 via 1360)
where the pH is raised to 10.5-12, preferably 11-11.5. In addition, clarified
raffinate 3110 can be sent to contractor 5000 for Mg removal. The process
chemistry is believed to be as follows:
Mg2+ + 504= + 2(R4N+01-11
-,E-phase
Mg(OH)2 particulate + ((R4N)2SO4)E-phase + purified waterpH 10.5-12
An emulsion comprised of Mg(OH)2 particulates, aqueous phase, and
extractant phase exits contractor 5000 via line 5010 and valve 5015 to line
5020 where the emulsion enters the optional 0/W separator 5030. The
emulsion exits 0/W separator 5030 via line 5035 and enters the solid/liquid
separator 5040. In the solid/liquid separator 5040, the Mg(OH)2 solids 5060
are separated from the emulsion via line 5065. The extractant phase is sent
via line 5050 to the third extraction operation 3020 for additional stripping
and regeneration. The pH 10.5-12 raffinate exits the solid/liquid separator
5040 via line 5070. The pH of the purified water can be reduced to the pH 6-9
range by sparging inline 5070 via valve 5075 with the CO2 gas 1371 via line
1372 and valve 6000. The pH 6-9 raffinate exits valve 5075 via line 5080 and
enters GAC 5085 for deodorization. The deodorized and purified water exits
GAC 5085 via line 5090 and is collected in a storage vessel 5100. The product
water is very pure, now being depleted of both M and N metals, Mn, Ca and
Mg, and with a 504= residual <20 ppm. This product water is of sufficient
quality as feed for most potable, industrial, and agricultural applications.
Table 1 lists examples of the water contaminants removed
simultaneously by the invention. Such contaminated water streams are often
so highly contaminated that no one technology is effective, or the contacting
element becomes fouled with other components of the mixture, or the water
64
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
is not sufficiently purified for release or reuse and must be treated again.
Such waters may contain many contaminants of concern under EPA
regulations. Such waters often include sulfate ion, a severe problem, in
concentrations of 150-2500 ppm and even higher.
Table 1. Typical water contaminants that can be removed by the invention
Class of Typical Contaminant Species Removed
Contaminants
ANIONS Sulfate and bisulfate, selenate, tellurate, phosphates
and hydrogen phosphates, organophosphates,
organophosphonates, organophosphinics,
polyphosphate (esp. ATP, ADP and AMP), arsenic
(especially arsenate, organ arsenates, and arsenite),
chloride, bromide, iodide, and pseudohalogen ions.
Nitrate, nitrite, cyanide ion, sulfide ion (H2S in
equilibrium with Hg, H2S and S'), oxometal ionic
complexes including single ion and molecular clusters
and colloids of molybdates (based on Mo042),
tungstates (based on W042), vanadates (based on
V032), chromates (based on HCr04-, Cr207, Cr042),
and the like, including their protonated weak acid
species, and particulate ion.
CATIONS Cations capable of forming oxide or hydroxide ion
colloids and precipitates ("oxohydroxo clusters or
colloids") including nickel, copper, chromium(III),
ferric, ferrous, aluminum, manganese, cobalt,
cadmium, zinc, Pb, Hg, Cd, and the like; and many
radioisotopes/ radioactive contaminants such as U, Th,
Pu, and the like hardness metal ions (Ca, Mg), and the
like contaminants such as Tc.
ORGANICS Oil-soluble organics, natural organic matter and the
like.
CA 02911119 2015-11-03
WO 2008/100610 PCT/US2008/002092
Below are analyzed samples of acid mine drainage from sites in Pennsylvania
(Tables 2A and 2B) and Ohio (Table 2C).
The Tables show chemical analysis of a actual representative
Pennsylvania acid mine drainage water samples to illustrate the extreme
contamination level that is not practically treated by any other known
technology. The stream in Table 2B is known to flow at about 10,000 gal/min
or more year after year resulting in many thousands of tons of metals and
sulfate contamination of the environment. It is unsuitable for potable water
production, for industrial process water use, for agricultural use, or for
1.0 providing aqua-tourism, or to support mountain stream life.
Table 2A. Analysis of Sugar Camp, PA Acid Mine Drainage Water
4841218/4841219 AMD Sample
Analysis Name Units Result MDL
Aluminum mg/I 2.73 0.0802
Calcium mg/I 128 0.104
Iron mg/I 204 0.0522
Magnesium mg/I 135 0.0135
Potassium mg/I 5.41 0.0503
Sodium mg/I 2.85 0.433
Cobalt mg/I 0.873 0.0021
Manganese mg/I 71.5 0.0036
Zinc mg/I 1.26 0.0081
pH 3.2 0.01
Std. Units
Alkalinity to pH 8.3 mg/I as N.D. 0.46
CaCO3
Alkalinity to pH 4.5 mg/I as N.D. 0.46
CaCO3
Total Dissolved Solids mg/I 2050 38.8
Sulfate mg/I 1620 60.0
Specific Conductance umhos/cm 2440 1.7
Acidity to pH 3.7 mg/I as 83.8 2.0
CaCO3
Acidity to pH 8.3 mg/I as 413 2.0
CaCO3
66
CA 0 2 911119 2 0 15-11-0 3
WO 2008/100610 PCT/US2008/002092
Table 2B. St.Michael, PA Acid Mine Drainage Water Analysis
Analysis Name St. Michaels AMD Feed Water 51944-01-18
Units Result MDL
=
Aluminum mg/I 4.16 0.0802
Calcium mg/I 158. 0.0632
Iron mg/I 169. 0.0522
Magnesium mg/I 160. 0.0135
Potassium mg/I 5.56 0.0503
Sodium mg/I 2.50 0.433
Cobalt mg/1 1.03 0.0021
Manganese mg/I 77.5 0.0042
Zinc 1.69 0.0081
pH 2.9 0.010
Std. Units
Alkalinity to pH 8.3 N.D. 0.46
mg/I as CaCO3
Alkalinity to pH 4.5 N.D. 0.46
mg/I as CaCO3
Total Dissolved Solids mg/I 2,310. 38.8
Sulfate mg/I 1,760. 60.0
Specific Conductance umhos/cm 2,720. 1.7
Acidity to pH 3.7 N.D. 10.0
mg/I as CaCO3
Acidity to pH 8.3 481. 10.0
mg/I as CaCO3
Table 2C. Rush Creek, OH High Al Acid Mine Drainage Water Analysis
Analysis Name High Aluminum AMD Feed Water 51944-24-06
Units Result MDL
Aluminum mg/I 133. 0.0802
Calcium mg/I 398. 0.0632
Iron mg/I 1,130. 0.261
Magnesium mg/I 151. 0.0135
Potassium mg/I 37.1 0_0503
Sodium mg/I 23.2 0.433
Cobalt mg/I 0.0758 0.0021
Manganese mg/I 28.7 0.0042
Zinc mg/I 0.970 0.0081
pH 3.0 0.010
Std. Units
Alkalinity to pH 8.3 N.D. 0.46
mg/I as CaCO3
Alkalinity to pH 4.5 N.D. 0.46
mg/las CaCO3
Total Dissolved Solids mg/1 7,350. 77.6
Sulfate mg/I 5,200. 150.
Specific Conductance umhos/cm 5,560. 1.7
Acidity to pH 3.7 N.D. 20.0
mg/I as CaCO3
Acidity to pH 8.3 2,290. 20.0
mg/I as CaCO3
N.D. = not detected
The invention is preferably used to treat the above mentioned waters
and the like before and/or instead of conventional waste water treatment to
obtain the maximum beneficial impact by
67
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
= Preventing the contamination of the environment and/or
= Pre-treating contaminated waters before these waters are treated in a
manner that generates large solid and/or liquid wastes thereby reducing the
total amount of solid and/or liquid waste produced.
This invention is particularly useful because it provides a broad
spectrum water purification capability for a wide range of water flow rates,
preferably flow rates of less than 1 to more than 10,000 gal/min.
Such extractant/co-extractant system is used alone or in combination
with one or more modifiers to improve extractant solubility in the diluent,
and/or with one or more water-immiscible diluents. Specific examples are
given in Table 3.
Suitable modifiers are water-immiscible terminal aliphatic alcohols or
mixtures thereof. Preferred diluents are alcohols that are classified
nonflammable (flash point > 140 F), nonhalogenated, low-odor, aliphatic,
either linear or branched, with a carbon number of 8 - 16, most preferably 9 -
13, or mixtures thereof. Specific examples are given in Table 3.
Suitable diluents can be water-immiscible aliphatic, aromatic solvents
or blends of such solvents. Most preferred are solvents that are classified
nonflammable (flash point > 140 F), nonhalogenated, low-odor aliphatic,
zo aromatic, or a blend of aliphatic and aromatic solvents. The aliphatic
diluent(s) can be linear but are preferably branched. The aromatic diluent(s)
can be unsubstituted aromatic liquids but are preferably aliphatically-
substituted aromatic liquid compounds. Extractant mixtures suitable for the
invention contain at least 25% diluent (v/v), preferably 60% (v/v), and most
preferably 85% (v/v). Specific examples of suitable diluents are shown in
Table 3.
For a further embodiment of the invention suitable reagents for
stripping of metal ions, regeneration, solute concentration, and/or pH
adjustment include mineral acids that do not cause decomposition of the
extractant. Suitable strip acids are preferably selected from hydrochloric,
sulfuric, phosphoric, blends of these and the like. Acid strength is 2-500/0
in
the feed solution and the S2M mixture is preferably maintained at - pH 1-2 or
68
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
less. Si mixer pH is preferably maintained at pH 2-4, or less. Sulfate ions
are subsequently stripped by basic reagents that do not cause decomposition
of the extractant preferably selected from alkali and alkali metal hydroxides,
and/or alkali carbonates. Base strength for sulfate ion stripping is
preferably
", pH 10-11 or greater. Specific examples of suitable of reagents for
stripping and regenerating, and pH adjustment of the extractant are listed
elsewhere herein.
69
CA 02911119 2015-11-03
WO 2008/100610 PCT/US2008/002092
Table 3. Typical Compounds Useful for the Extractant Phase
Chemical
Class (used
Extractant
alone or in Level of
Formulation Specific Compounds
combination Preference
Component
with any other
extractant)
N-methyl tri-(n- Most preferred
octyl)ammonium ion
N-methyl tri-(n- Most preferred
decyl)ammonium ion
N-methyl tri-(n- Most preferred
dodecyl)ammonium ion
Aliquat 134 Most preferred
Aliquat 336 Most preferred
Tri-octyl methylarnmonium ions Most preferred
Quaternary Mixture of tridecyl- and trioctyl- Most preferred
Amines methylammonium ions
Extractant HOE S 2706 Most preferred
Adogen 464 Most preferred
Tri(C8-C10) methylammonium ions Most preferred
RI=R2=R3=CH3(CH2)9- Most preferred
RIR2R3N+CH3 R1=R2=le=CH3(CH2)7- Most preferred
and Most preferred
RI=R2=R3=C113(CH2)9-
Blends of the above quaternary Most preferred
ammonium ions in any proportion
Mono Most preferred
e.g. LDC-79
guanadinium
Quaternary
N-methyl tri(n-octyl) phosphonium Functional
phosphonium
b-isodecanol Most preferred
(or decyl
alcohol, or
Exxal 10)
Isotridecanol Most preferred
(orb-
Modifier
Tridecyl
alcohol
Nonyl phenol aromatic Functional
Dodecyl aromatic Functional
phenol
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Table 3. (Continued)
Extractant Chemical Specific Compounds Level of
Formulation Class (used Preference
Component alone or in
combination
with any other
extractant)
Aromatic Functional
150
Aromatic0 Functional
200
Diluent
Calumet Most Preferred
400-500
Conoco 170 Preferred
Isopare M Preferred
Referring to Table 3, effective "Type extractant phase" extract
compounds of the invention. All of the components of the E-phase are oil
soluble with a total carbon number of at least eight (8), can be charged or
neutral, and can have additional functional groups such a halogens, ether
linkages, ester linkages, alkyl phenolic, and the like, so long as the
extraction
chemistry and the oil solubility of the reagent is not adversely effected
relative to the descriptions herein.
Effective extractant compounds of the invention are all oil
soluble with a total carbon number of at least eight (8), but preferably about
16, and most preferably about 25 or more, and can have additional functional
groups such a halogens, ether linkages, ester linkages, aromatic groups, be
linear or branched, blends of these, and the like, so long as the extraction
chemistry and the oil solubility of the reagent is not adversely effected.
Referring now to Figure 1A and Figure 18, a general overview of the
process is shown. The figure shows extractant phase continuous recycle and
operation at pH up to about 11.0-12.0 (Mn and Mg recovery). Thus
contaminated water (e.g. acid mine drainage) is treated with extractant phase
and within a short time water, together with solid products, is moved to a
solid/liquid separation apparatus where metal oxide/hydroxide carbonate
products {e.g. MnCO3 (Ca CO3), Mg(OH)2} are removed. The water then
flows to an oil/water separator that provides a purified water product. Other
71
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
contaminants are removed with LLX extraction in a loaded extractant phase
that produces a floc. In addition to LLX extraction, product recovery and
extractant phase regeneration, stripping is used to remove other metal
oxide/hydroxide/ carbonate products (e.g. Fen, Fe", Al, Ni, Co, and Zn) that
were separated in the additional floc using H2SO4 and (Na/K)2CO3, typically
enables these metal sulfate products to be recovered.
Alternate liquid-liquid contacting apparatus can be provided as mixer-
settler, columns, in-line mixers, contacting centrifuges, and the like.
Referring now to Figure 3, one embodiment comprises four (4)
extraction stages for metal cation and sulfate anion co-extraction, two (2) or
four (4) acid stripping stages for metal stripping (depending on whether
metals are to be separated during production of metal concentrates), and four
(4) stripping stages for sulfate stripping and extractant regeneration. The
overall process flow diagram using a combination of conventional and
uniquely designed mixer-settlers is shown in Figure 3.
The flow configuration of the device of Figure 3 enabled the steady
flow of heavy floc that was produced in El settler 316 to flow to the Sl-M 351
(shown as Sl-MS04) stripper, where the floc is converted back to a two phase
liquid form manageable by conventional LLX hardware. The floc flow in El
312, E2 313, and the associated decanter 314 is accomplished by the "T"
shaped configuration along with the floc flow handling weirs contained in the
settlers and at the overflow point. These weirs are described elsewhere in
this application. Note that with the "T" design, the E-phase overflow from E2
settler 317, representing 325b, the more flowable floc shears off the heavy
floc exiting from El settler 316, and hence insures that the floc from El
settler 316 flows steadily. The E-phase flow rates to El and E2 are adjustable
and are used to optimize floc flow continuity, extraction yield, and reagent
consumption minimization.
Also Figure 3 illustrates the wraparound (space saving) means to
increase the residence time in the settlers of E3 341 and E4 342. Enhanced
residence time in these settlers is desirable as the total dissolved solids
level
in the purified water decreases and becomes slower to phase separate. As
72
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
before, all of the strippers of the "T" configuration of Figure 3 are
preferably
operated counter-current with most preferred internal recycle of aqueous.
Note that the extractors were operated cross-current with respect to the E-
phase flow, and where the E3 341 and E4 342 stages are operated counter-
current. Each loaded E-phase could have been stripped separately, but they
are shown recombined in Figure 3. Keeping these three loaded E-phases
separate during stripping would allow production of three separate metal ion
products (see Detailed Description of the Invention).
Referring again to Figure 3, the disclosed process uses a floc liquid-
liquid extraction system to extract metal ions and sulfate ions. This
typically
is useful for cleaning up a contaminated and environmentally harmful acid
mine water discharge stream. Due to the unique nature of the acid mine
drainage stream and the floc-based process chemistry, the extraction system
apparatus has design requirements that differ from conventional liquid-liquid
mixer-settler systems.
Conventional mixer-settler based LLX systems have the advantage of
self-regulating and low maintenance and labor cost. However, the
conventional mixer-settler system performs poorly when dealing with metal
oxide colloids and/or floc, and would easily completely fail in minutes with
the
zo acid mine drainage feed water discussed here. A driving force for one
aspect
of the present invention is,to change the conventional LLX contactor design to
create a new way of dealing with the metal oxides/hydroxide colloids, flocs,
particulates or slurry while retaining the advantages of conventional mixer-
settler systems. This new design handles flocs along with liquid-liquid
processing, or flotation liquid-liquid extraction.
Referring again to Figure 3 that illustrates one embodiment according
to the invention as applied to acid mine drainage water cleanup using the
simultaneous extraction process of the invention in a specially designed
apparatus 300. This apparatus 300 typically has a acid mine drainage (AMD)
water inlet 301 and various interconnections for liquid flow referred to as
internal recycle lines 303, aqueous lines 304, and extractant lines 305. The
water 301 flows into a T shaped extraction decanter apparatus 311 made up
73
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
of a first extractor El 312, a second extractor E2 313, and a dual decanter
314. The extractor El 312 has a mixer 323 which receives the incoming
water 301 via an aqueous line. Here the water 301 is mixed with extractant
solution 302 (the extractant solution is loaded with an anionic base - in this
case sodium carbonate)from the extractant solution storage tank 307 via an
extractant line and pump 318. The mixture flows into the settler section 316
of extractor El 312 where the extractant 302 and water 301 interact to form
a floc. The floc is allowed to separate typically by being allowed to float to
the surface, a flow guide 324a helps the floc to flow over a floc weir 325a.
The floc containing extractant, some water and captured ions flows over the
floc weir 325a into the dual decanter 314. The aqueous portion of the
material left behind in El settler 316 flows via an aqueous line to the mixer
331 of extractor E2 313. Additional extractant solution 302 from tank 307
flows via an extractant line and pump 319 to mixer 331 where the water from
El settler 316 is mixed with the additional extractant solution 302 which then
flows into the settler section 317 of extractor E2 313. After also being
allowed to form a floc and separate from most of the aqueous the floc is
guided by flow guide 324b over the floc weir 325b and into the dual decanter
314. The flocs from El settler 316 and E2 settler 317 typically mix and are
allowed to separate from entrained aqueous. The newly formed and/or
reformed floc in the dual decanter 314 is guided by a flow guide 324c to flow
over the floc weir 325c into a collector where the floc flows to the first
stripper 91-MS04 351.
Aqueous depleted in ions in dual decanter 314 is typically withdrawn
from the bottom and flows via aqueous line 330 to extractor E3 341 where it
enters at mixer 332, here extractant from an extractant line 331 from
extractor E4 342 also enters the mixer 332. After mixing the aqueous and
extractant phases, the mixture enters the settler 333 of extractor E3 341.
The mixture is allowed to separate into an aqueous phase and an extractant
phase. The extractant phase is withdrawn by an overflow weir into a collector
334 having an outlet 335 and then flows via an extractant line into the mixer
352 of 91-M904 stripper 351. Aqueous flows to outlet 336 and via an
74
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
aqueous line to the mixer 343 of extractor E4 342. In addition extractant
solution 302 is pumped via pump 306 to mixer 343. After mixing with
aqueous from outlet 336 of extractor E3, the mixture flows through the settler
345 of the extractor E4 342. The mixture is allowed to separate into an
aqueous phase and an extractant phase. The extractant phase is withdrawn
by an overflow weir into a collector 344 having an outlet 345 and then flows
via an extractant line into the mixer 332 of extractor E3 341. Aqueous leaves
extractor E4 342 via outlet 346 to an oil water separator 347 and then to a
sand filter 348 to obtain purified water 349.
Sulfate is stripped from the extractant in strippers 366, 371, 376, 381.
Strippers 351 and 356 serve to remove metal ions with a charge of about +3
as metal sulfate product 355. Extractant, floc and entrained aqueous also
flow to the mixer 352 of stripper 351 from the outlet 315 of dual decanter
314. Inflow of extractant to mixer 352 mixes with aqueous inflow from
stripper 356 and the mixture in stripper 351 is separated into an aqueous
MS04 product 355 and an extractant that flows to a mixer 357 in stripper
356. Note that all the strippers have internal recycle lines 303 for aqueous
recycle flow. The incoming extractant from stripper 351 is mixed in mixer 357
with sulfuric acid 353 (in this example about 50%) to make the mixture have
zo a very low pH so that the metal is stripped from the extractant and
driven
into the aqueous phase. Extractant leaves stripper 356 and flows via an
extractant line 305 to mixer 367 of sulfate stripper 366. In mixer 367
aqueous flow from stripper 371 mixes with the incoming extractant from
stripper 356. Sodium sulfate 368 that is a very useful product flows from the
outlet of stripper 366. An extractant line 305 from stripper 366 provides for
flow of extractant to the mixer 372 of stripper 371. Aqueous from stripper
376 flows to the mixer 372 and is mixed with the incoming extractant from
stripper 366. Extractant further stripped of sulfates exits stripper 371 and
flows to the mixer 377 of stripper 376, here it is mixed with incoming aqueous
from stripper 381. Extractant further stripped of sulfates exits stripper 376
and flows to the mixer 382 of stripper 381, here it is mixed with incoming
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
carbonate 383 and the like. The extractant essentially stripped of metals and
sulfates flows from stripper 381 to an extraction solution storage tank 303.
Referring now to Figure 4, the present invention provides some
designed features that are specific to assist flowing and maintaining the flow
of the metal oxide/hydroxide colloids, flocs and particulate slurries. The
detailed schematic of Figure 4 shows certain elements of the designed
flotation device to assist in maintaining the flow of extractant phase over
the
settler overflow weir while not interfering with aqueous phase clarification.
The flotation device flow pattern is also illustrated to visualize the
movement
of the metal ion colloids, flocs and/or particulate slurry.
The apparatus shown in Figure 4 represents a typical mixer-settler
configuration. The apparatus has a mixer compartment 404 that has a
rotating mixer impeller 402 which creates a suction that pulls the AMD water
and extractant phase 406 into the mixer compartment 404 where it mixes the
Is AMD water and extractant phase 406 thoroughly. The mixing forces the
newly
formed aqueous-extractant phase emulsion 424 over the top of the mixer
compartment 404 and under the underflow weir 409 into the settler
compartment 420. Here the mixed phase emulsion 424 is allowed sufficient
time to break and split into 2 separate phases. The upper layer of the settler
compartment 420 contains the extractant phase with floc and ions 422. The
lower layer contains the aqueous phase 426 that has been treated and
depleted of ions. The bottom of the settler compartment 420 contains an
outlet 438 for the aqueous phase.
At the end of the settler compartment 420, there is a floc weir 430 that
facilitates the extractant-floc phase movement. This weir contains a floc or
slurry entrance surface positioned at typically about 300 from the vertical
angle. The smooth entrance ramp 432 and exit ramp 434 provide the least
resistance for the extractant phase-floc phase to move over the surface of the
floc weir 430. And what is more, this angle of floc approach and exit,
combined with the length of the ramps to use the inherent internal colligative
property of the floc, which is gel-like in consistency, to enable the exiting
floc
to literally pull the entering floc over the smooth weir. In this manner, floc
76
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
flow is maintained continuous or semi-continuous. Literally, the weight of the
falling floc film and associated extractant phase pulls the entering
extractant-
floc phase layer to and over the weir. The stronger the internal colligative
strength the shorter the ramp can be for a given fluid viscosity. Both an
entrance angle and an exit angle are important design parameters as shown
in the figure. Typically both of the angles can be the same or different and
can be from about 100 to about 80 , more preferably from about 15 to
about 45 . At the right hand side of the figure, the rounded lip 436 of the
floc weir 430 enables efficient flow over the weir while the rounded bottom
435 of the floc weir 430 enables reliable gravity-driven break-off of the
exiting
floc containing extractant phase liquid film that creates a flow pattern that
allows the overflowing extractant phase to be dripped into the extractant
solution where it exits the device via the extractant trough or launderer to
the
next stage. Since the controls are typically performed via gravitational
force,
the drip point 437 is at the lowest point of the floc weir exit slide and the
metal ion colloid thickened phase will not linger or attach to the sides of
the
weir or the tank.
An optional standpipe 450 that may be adjustable in height may be
used to control the outflow of aqueous. The standpipe 450 may be internal
to the settler compartment 420 as shown in Figure 4 may be external as is
known in the art (not shown).
Referring now to Figure 5, a further component of the invention is a
extractant phase floc flow guide for the settler and/or decanter. The upper
right hand corner of the drawing shows a 3-dimensional view of a block
shaped flow guide that is also adjustable, it may be made of different layers
of flat stock fixed on top of each other for thickness tunability of the block
to
allow optional flow control for different types and thicknesses of floc or may
be a solid block. The device is hereinafter called a flow guide. The flow
guide may be in the form of a block shaped flow guide 510a or as a plate
shaped flow guide 510b. For further discussion below, the designation flow
guides 512a, 512b, 512c will be used.
77
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Figure 5 also shows an expanded version of the extractor decanter
system from Figure 3 but the larger scale showing more detail. The view is
from the top and shows the flow guides installed into the "T" designed mixer-
settler described above (Figure 3). When acid mine drainage water enters
s inlet 502 of mixer 514 in extractor 508 it is mixed with extractant
solution and
flows over or through a weir 511 to a settler 516. The flow guide 512a aids
in maintaining the flow of extractant phase-floc (typically metal ion,
hydroxide, sulfide containing) as it thickens during its travel along the
settler
516 and over a floc weir 517 where the floc exits the settler 516 and flows
into a decanter 530. Aqueous and extractant entering the inlet of mixer 524
of extractor 520 is mixed and flows over or through a weir 513 into a settler
526. The flow guide 512b aids in maintaining the flow of extractant phase-
floc (typically metal ion, hydroxide, sulfide containing) as it thickens
during its
travel along the settler 526 and over a floc weir 527 where the floc exits the
Is settler 526 and flows into a decanter 530. The inflowing floc from floc
weir
517 and floc weir 527 combine in decanter 530 further separating into an
extractant phase containing floc and aqueous phase. The flow guide 512c
aids in guiding and maintaining the flow of the relatively thick floc over the
floc weir 537 to outlet 540. The aqueous outlet pipes 519a, 519b, and519c
provide individual control of aqueous phase level and floc thickness in
settler
516, settler 526 and decanter 530 typically by adjustment of the height of the
respective pipe. The flow guides 512a, 513b, 513c are placed to have an
underflow channel not visible in this top view, see Figure 5A for a side view.
Thus the aqueous phase flows under the flow guides that simultaneously
block the flow of extractant phase and floc to the aqueous outlet pipes 519a,
519b, 519c. The arrows show typical liquid flow directions.
Figure 5 shows the geometry of the flow guides and where the flow
guide is located in the settler or decanter. The flow guide is designed to
narrow the settler liquid flow channel most preferably only near the top
surface of the settler so that it only increases the velocity in the floc-
loaded
extractant phase and thus help in moving and for thickening the metal oxide
colloids that are the floc and are located above the water phase. The flow
78
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
guide combines with the flotation overflow weir device described above, to
dramatically increase the flow of metal colloids floc particulates extractant
phase into the next mixing chamber. The flow guide, which can also be
constructed as just as an internal wall or partition that is raised at the
bottom
to allow the aqueous phase to under-flow it to allow continuous removal of
aqueous layer via a suitable weir sump or stand pipe. Alternatively the flow
guide can be substituted with a thin plate of material as shown.
Figure 5A shows a side view of the mixer 524 and settler 526 of Figure
5. It shows the geometry and location of the flow guide 512b in settler 526.
The extractant phase and AMD water 522a are pulled into the mixing
compartment 524 by the suction of the rotating mixer impeller 524. In the
mixer compartment 524, the AMD water and extractant phase 522a mix
thoroughly. The mixing forces the newly formed aqueous-extractant phase
emulsion 527a over the top of the mixer compartment 524 (in some
embodiments the flow is under a weir 513) into the settler 526. In the settler
526, there is a flow guide 512b which moves the extractant phase and floc
525 along to the next mixing chamber. The figure shows the flow guide 512b
constructed as an internal wall partition that is raised at the bottom to
allow
space 512b3 for the aqueous phase 522c to underflow the flow guide 512b to
zo allow continuous removal of aqueous phase522c via a stand pipe 519. Floc
522b flows out over the floc weir 527
Figure 5B shows an alternative version of Figure 5A using a block 510a
as a flow guide. In this embodiment, the flow guide 512b consists of an upper
channel narrowing block located in the settler 526. The block shaped flow
guide 512b is adjustable and it may be made of different layers of flat stock
fixed on top of each other for thickness tunability of the block 510a to allow
optional flow control for different types and thicknesses of floc or may be a
solid block. In this figure, there is an opening 519b at the bottom of settler
526 which allows for the removal of aqueous phase 519c. Floc 522b flows
over the floc weir 527.
Figure 5C shows the flow configuration of the El mixer compartment
of Figure 17. The extractant phase and floc 541 from E2 decanter flow over
79
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
the floc weir 540 into the extractant trough or launderer 542. The extractant
phase and floc 541 are pulled into the mixing compartment 544 along with
the incoming AMD water 543 by the suction of the rotating mixer impeller
545. In the mixer compartment 544, the AMD water 543 and extractant phase
541 mix thoroughly. The mixing forces the newly formed aqueous-extractant
phase emulsion 547 over the top of the mixer compartment 544 and under
the underflow weir into the El settler compartment 546.
Gravity Benefits
lo Depending on the geographical location of the process unit, the water
treatment process according to the invention could be arranged in series to
take advantage of the slope of a hill and make the gravitational flow more
efficient by providing steeper descents and deeper extractant phase liquid
thickener at over-flow weirs without the need to construct such gradients.
Stage-to-Stage Loaded Extractant Phase Transfer Designs
When the unit extraction contactors configurations are in series, there
is an advantage to introduce the extractant solution without passing the floc
through piping to the mixer from the bottom. Though both floc-loaded
.. modes work, most preferred is to avoid sending the floc through smaller
I.D.
piping to accomplish stage to stage transfers. This feature avoids having the
thickened extractant phase slurry or floc flow through piping and thereby run
the risk of pipe or drain pluggage. However, when the extract material is
introduced into a mixer (e.g. S1-M from ElD) from the top, prevention of
extractant flowing over into the stripper settler without proper mixing is
critical to prevent short circuiting of the extractant phase flow. Sufficient
mixing time of the floc in the strip mixer is required to allow enough time
and
access to aqueous strip acid to dissolve the colloid/floc back into a
conventional metal ion sulfate solution. Only enough time is needed to break
the flocs affiliation with E-phase as continued hydrolysis reactions can
continue in the internally recycled aqueous phase. This short circuiting is
prevented by the trough or hopper feeding of the floc feed into the manifold
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
beneath the mixer which feeds flocks, slurries, and fluids to the mixing
compartment by suction from the impeller. These designs that are discussed
below are readily implemented and do not require complex parts.
Referring now to Figure 6, the most preferred design for stage-to-stage
floc transfer is to utilize an underflow weir 409 similar to the one that is
separating the mixer compartment 404 and settler compartment 420. Figure
6 shows the underflow inlet weir 408 position for introducing extractant floc
solution 422 from the top of the mixer compartment 404 and without piping.
The rotating mixer impeller 402, while in action, creates a suction that pulls
io freshly added floc-loaded E-phase 422 to the mixer 402, where it mixes
the
extractant solution 422 and inlet aqueous phase 426 thoroughly while the
underflow weir 409 prevent premature exit of the extractant 422 across the
top of the mixer compartment 404 without first being thoroughly mixed with
the inlet aqueous phase 426.
Referring now to Figure 7, another design, according to another aspect
of the invention, is to place an extractant solution introduction inlet 480
located inside the mixer compartment 404. The inlet 480 achieves a similar
effect as the underflow weir 409 by insuring thorough extractant solution 422
and aqueous phase 426 mixing and reactor residence time. In addition, the
zo bend elbow design on the bottom of the extractant phase inlet line 480
(piping or tube) prevents feed water 426 back-flowing into the tube which
can avoid problems due to solids formation and potential pluggage. The
rotating mixer impeller 402 is most preferably designed to create a suction
that brings the extractant solution 422 to the bottom of the mixing
compartment 404 and thereby promotes thorough mixing with feed water
426.
Referring now to Figure 8, another preferred design useful to enable
processing of extractant phase flocs and slurries when introducing extractant
solution with minimum piping is to include a storage chamber 442 before the
mixing compartment 404 and to introduce the extractant solution 422 from
the bottom of the mixing compartment 404 instead of from the top. The
inverted "y" shaped design feature at the bottom of the storage chamber 442
81
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
minimized aqueous phase 426 back-flowing into the extractant storage
chamber 442 and thereby avoids slurry pluggage of the extractant phase inlet
line. This design also provides some inline contact between extractant 422
phase and aqueous phase 426.
Figures 9, 10, 11, and 12 provide typical extraction and stripping
McCabe-Thiele diagrams that were measured experimentally for candidate
extractant formulations. These plots enable the user to determine the best
mode of operation of the invention with respect to the optimal E/A ratios and
the number of stages needed for both the extraction and stripping of sulfate
ions. These plots also identify the concentration of the sulfate product
produced and the level of residual sulfate remaining in the purified water at
a
selected set of operating conditions.
Referring now to Figure 13, the embodiment of the sulfate circuit
comprises four extraction stages for metal cation and sulfate anion co-
extraction and four stripping stages for sulfate stripping and extractant
regeneration. The overall process flow diagram is shown in Figure 13.
The apparatus has a Na2SO4 solution (2000ppm) inlet 1300 and various
interconnections for aqueous flow referred to as internal recycle lines 1381,
aqueous lines 1383, and extractant lines 1385. Sulfate is stripped from the
extractant in strippers S1-SO4 1339, S2-SO4 1338, S3-SO4 1337, and S4-SO4
1336. In addition, the extractant phase is regenerated in the strippers. Note
that all the strippers have internal recycle lines 1381 for aqueous recycle
flow.
Aqueous phase is depleted of ions in El decanter 1340 and extractors El
1341, E2 1341, E3 1343, and E4 1343.
The Na2SO4 solution flows via pump 1315 and line 1302 to the mixing
compartment of the first extractor El 1341. In addition, extractant phase and
emulsion from extractor E2 1342 flows to extractor El 1341 via line 1306.
The extractant phase and emulsion exits extractor El 1341 via line 1303 and
enters El decanter 1340. In the decanter, the emulsion is allowed enough
time to break and separate into an aqueous and extractant phase. The
extractant phase flows to S1-SO4 1339 via line 1305 while the aqueous is
withdrawn and sent to extractor El 1341 via line 1322. The extractant phase
82
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
flows into S1-SO4 1339, exits via line 1307, and enters S2-504 1338. The
aqueous phase exits S1-SO4 1339 as the Na2SO4 concentrate product 1350
via line 1328. The extractant phase flows into S2-SO4 1338, exits via line
1313, and enters S3-SO4 1337. The aqueous phase exits S2-SO4 via line 1309
and flows into S1-SO4 1339. The extractant phase flows into S3-SO4 1337,
exits via line 1315, and enters S4-SO4 1336 along with the incoming
carbonate solution 1340 which flows via pump 1335 and line 1319. The
aqueous phase exits S3-SO4 via line 1311 and flows into S2-SO4 1338. The
extractant phase flows through S4-504 1336, exits via line 1321, and returns
.. to the regenerated extractant solution tank 1360. The aqueous phase leaving
S4-504 1336 exits via line 1317 and flows into S3-SO4 1337.
The aqueous phase withdrawn from El decanter 1340 flows into
extractor El 1341 via line 1322. The extractant phase leaving El decanter
1340 exits via line 1305 and flows into Sl-SO4 1339. The aqueous phase
flows through El 1341,exits via line 1304, and flows into extractor E2 1342.
The extractant phase exits El 1341 via line 1303 and flow into El decanter
1340. The aqueous phase flows through E2 1342, exits via line 1310, and
flows into extractor E3 1343. The extractant phase leaving E2 exits via line
1306 and flows into El 1341. The aqueous phase flows through E3 1343,
zo exits via line 1312, and flows into extractor E4 1344. The extractant
phase
exits E3 1343 via line 1310 and flows into E2 1342. The aqueous phase flows
through E4 1344, exits via line 1318, and flows into the 0/W separator 1380.
The extractant phase exits E4 1344 via line 1314 and flows into E3 1343. The
purified water 1370 exits the 0/W separator 1380 via line 1320. The
.. extractant phase in the 0/W separator 1380 exits via line 1324 and flows
into
E4 1344 along with aqueous phase from E3 1343 which flows via line 1312
and additional extractant phase 1360 which flows via pump 1325 and line
1316.
Referring now to Figure 14, this figure illustrates acid mine drainage
water purification process flow Sheme 1. Process flow Scheme 1 provides the
ability to separate the metal ion components of the acid mine waters into
separate products as desired. Figure 14 illustrates how this is accomplished
83
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
for two groups of metal ions, M and N. Given this information, one skilled in
the art would be readily able to proform more that two separations by adding
additional extraction or metal ion strip circuits. This capability is
described as
follows using Figure 14 which deploys process flow scheme 1. More than two
extraction stages could be used for each extraction or strip circuit, but two
were found sufficiently effective in the current invention for most needs,
however three or four stages are preferred for each extraction or strip
circuit.
Figure 14 illustrates Scheme 1 and comprises two extraction stages for
M metal cation and sulfate anion co-extraction, two extraction stages linked
in
series to the first two stages for N metal cation and further sulfate anion co-
extraction, two (preferably three) acid stripping stages for M-metal stripping
and two (preferably three) acid stripping stages for N-metal stripping ,and
four (preferably 4 to 6) stripping stages for sulfate stripping with
simultaneous extractant regeneration.
The apparatus has an AMD feed water inlet 1400 and various
interconnections for aqueous flow referred to as internal recycle lines 1483,
aqueous lines 1481, and extractant lines 1482. Sulfate is stripped from the
extractant in strippers S1-SO4 1413, S2-SO4 1412, 53-SO4 1411 and 54-SO4
1410. In addition, the extractant phase is regenerated in the strippers. Note
zo that the metal and sulfate strippers have internal recycle lines 1483
for
aqueous recycle flow. Aqueous phase is depleted of ions in E1-MS04
Decanter (D) 1416, E1-NS04 Decanter (D) 1424, and extractors E1-MS04
1418, E2-MS04 1419, E1-NS04 1420, and E2-NS04 1422.
The AMD feed water 1400 flows via pump 1475 and line 1403 into El-
MS04 mixer 1418 along with extractant phase and floc from E2-MS04 via line
1425. The aqueous phase (raffinate) exits E1-MS04 1418 via line 1444 and
flows into E2-MS04 1419. The extractant phase exits E1-MS04 1418 via line
1426 and flows into E1-MS04 Decanter (D) 1416. The aqueous phase leaving
E1-MS04 Decanter (D) 1416 flows into the raffinate return 1485 via line 1441.
Raffinate Return from any Decanter of the invention represents a relatively
small amount of aqueous phase flow that corresponds to that volume of
physically entrained aqueous phase that accompanied the extractant phase
84
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
flow as the latter exited its extractor (in this case E1-MS04). It is an
important and unique feature of the floc based liquid liquid extraction
technology that such physically entrained aqueous flows exist. These small
aqueous flows exist due to the requirement that the floc not be allowed to
s thicken excessively in the extracter to avoid it thickening too much
there and
eventually solidify and plug up the setter and thereby becoming retained by
the extractor. This problem is avoided by the technology by operating such
to keep the extractant phase layer thin in each extracter so as to reduce the
residence time of the extract layer short. The thin nature of the extractant
phase then allows some co-flow of aqueous layer as the mixture approaches
the floc over flow weir. This aqueous flow is easily collected in the decanter
and recycled as shown in the figures.
The extractant phase leaving E1-MS04 Decanter (D) 1416 flows into
S1-MS04 mixer 1415 via line 1427. The aqueous phase exits S1-MS04 1415
as the M-metal sulfate product (MS04) 1491 via line 1440. The extractant
phase exits S1-MS04 1415 via line 1428 and flows into S2-MS04 mixer 1414
along with the 50 wt% sulfuric acid solution 1490, (or other strip acid feed
which is 2-70% in concentration, and preferably 15 to 50% concentration),
which flows via pump 1474 and line 1405. The aqueous phase leaving S2-
MS04 1414 flows into S1-MS04 1415 via line 1439. The extractant phase
leaving S2-MS04 1414 flows into S1-SO4 1413 via line 1429 along with
aqueous phase from S2-SO4 1412 which flows via line 1436 and extractant
phase from S2-NS04 1422 which flows via line 1456. The aqueous phase
exits S1-SO4 1413 as the Na2SO4 product 1489 via line 1437. The extractant
phase exits S1-SO4 1413 via line 1430 and flows into S2-SO4 mixer 1412
along with aqueous phase from S3-SO4 via line 1435. Carbon dioxide (CO2)
1486 exits S1-504 1413 via line 1438, which is optionally captured and used
as a co-product or vented. The aqueous phase leaving S2-SO4 1412 flows
into S1-SO4 mixer 1413 via line 1436. The extractant phase leaving S2-SO4
1412 flows into S3-SO4 mixer 1411 via line 1431 along with aqueous phase
from S4-SO4 1410 which flows via line 1434. The aqueous phase exits S3-
SO4 1411 and flows into S2-SO4 mixer 1412 via line 1435. The extractant
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
phase exits S3-SO4 1411 and flows into S4-SO4 mixer 1410 via line 1432
along with 1 to 25% Na2CO3 solution, preferably the 10 to 20% Na2CO3 , and
most preferably 13 -17% Na2CO3solution 1488 which flows via pump 1473
and line 1406. The aqueous phase leaving S4-SO4 1410 flows into S3-SO4
mixer 1411 via line 1434. The extractant phase leaving S4-504 1410 returns
to the extractant solution storage tank 1487 via line 1433.
The aqueous phase leaving E1-MS04 1418 flows into E2-MS04 mixer
1419 via line 1444 along with extractant phase from the extractant storage
tank 1487 which flows via pump 1472 and line 1401. The extractant phase
io leaving E1-MS04 1418 flows into E1-MS04 Decanter (D) 1416 via line 1426.
The aqueous phase exits E2-MS04 1419 and, for the case of using only two
"M" extraction stages, flows into E1-NS04 stage 1420 via line 1445 along with
extractant phase from E2-NS04 1421 which flows via line 1448. The
extractant phase exits E2-MS04 1419 and flows into E1-MS04 1418 via line
1425. The aqueous phase leaving E1-NS04 1420 flows into E2-N504 mixer
1421 via line 1446. The extractant phase leaving E1-NS04 1420 flows into
Decanter (D) 1424 via line 1453. The aqueous phase exits E2-NS04 1421 via
line 1447 and flows into 0/W separator 1417. The 0/W separator 1417
effluent exits via line 1443 as the purified water product 1493. Depending on
the requirements for use or environmental release of this water it can be
released, deodorized, and/or filtered for solid particulate removal. The
recovered low flow of extractant phase exits the 0/W separator 1417 via line
1442 and flows into the extractant solution recycle tank 1492. The aqueous
phase leaving Decanter (D) 1424 flows to the raffinate return 1484 (see
.. above for definition of Raffinate Return) via line 1454. The extractant
phase
leaving Decanter (D) 1424 flows into S1-NS04 1423 via line 1452 along with
aqueous phase from S2-NS04 1422 which flows via line 1149. The aqueous
phase exits S1-NS04 1423 via line 1451 as the N-metal sulfate product 1495.
The extractant phase exits S1-NS04 1423 via line 1450 and flows into the S2-
NS04 mixer 1422. The stripped extractant phase from S2-NS04 1422 settler
and flows to the S1-SO4 sulfate stripper 1413 via line 1456.
86
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Referring now to Figure 15, this figure illustrates acid mine drainage
process flow scheme 2. Process flow scheme 2 comprises four extraction
stages for metal cation and sulfate anion co-extraction, two acid stripping
stages for metal stripping, and four stripping stages for sulfate stripping
and
s .. extractant regeneration. The overall process flow diagram is shown in
Figure
15.
The apparatus has an AMD feed water inlet 1500 and various
interconnections for aqueous flow referred to as internal recycle lines 1591,
aqueous lines 1592, and extractant lines 1593. Sulfate is stripped from the
3.0 extractant in strippers S1-SO4 1539, S2-SO4 1538, S3-SO4 1537, and S4-
SO4
1536. In addition, the extractant phase is regenerated in the strippers. Note
that the metal and sulfate strippers have internal recycle lines 1591 for
aqueous recycle flow. Aqueous phase is depleted of ions in El Decanter (D)
1542 and extractors El 1543, E2 1544, E2 Decanter (D) 1545, E3 1546, and
15 E4 1547.
The AMD feed water 1500 flows via pump 1555 and line 1530 into El
mixer 1543 along with extractant phase and floc from E2 1544 via line 1525
and aqueous phase from ElD 1542 via line 1524. The aqueous phase exits
El 1543 via line 1526 and flows into E2 mixer 1544. The extractant phase
zo .. exits El 1543 and flows into ElD mixer 1542 via line 1507. The aqueous
phase leaving E1D 1542 flows back to El mixer 1543 via line 1524. The
extractant phase leaving ElD 1542 flows to Sl-MS04 mixer 1541 via line
1508 to line 1535 along with extractant phase from E2D 1545 via line 1506
and extractant phase from 0/W separator 1598. The aqueous phase exits
25 S1-M504 1541 as the metal sulfate concentrate product 1596 via line
1548.
The extractant phase exits S1-MS04 1541 via line 1509 and flows into S2-
MS04 mixer 1540 along with the 50 wt% sulfuric acid solution which flows via
pump 1585 and line 1532. The aqueous phase leaving S2-MS04 1540 flows
to Sl-MS04 mixer 1541 via line 1523. The extractant phase leaving S2-MS04
30 .. 1540 flows to S1-SO4 mixer 1539 via line 1510. The aqueous phase exits
Sl-
504 1539 as the Na2SO4 product 1592 via line 1533. The extractant phase
exits Sl-SO4 1539 and flows into 52-SO4 mixer 1538 via line 1511. The
87
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
aqueous phase leaving 52-SO4 1538 flows into S1-504 mixer 1539 via line
1522. The extractant phase leaving S2-504 1538 flows into S3-504 mixer
1537 via line 1512. The aqueous phase exits S3-504 1537 and flows into S2-
504 mixer 1538 via line 1521. The extractant phase exits S3-504 1537 and
flows into 54-SO4 mixer 1536 via line 1513 along with the 15 wt% Na2CO3
solution which flows via pump 1595 and line 1534. The aqueous phase
leaving S4-504 1536 flows to 53-504 mixer 1537 via line 1520. The
extractant phase leaving S4-504 1536 is regenerated and returns to the
extractant solution storage tank 1597 via line 1514.
The aqueous phase withdrawn from ElD 1542 via line 1524 flows into
the El mixer 1543. The aqueous phase exits El 1543 via line 1526 and flows
into E2 mixer 1544 along with the extractant solution from the storage tank
1597 which flows via pump 1575 and line 1531. The extractant phase exits
El via line 1507 and flows into ElD mixer 1542. The aqueous phase leaving
is E2 1544 flows into E3 mixer 1546 via line 1527 along with aqueous phase
from E2D 1545 via line 1528 and extractant phase from E4 1547 via line
1504. The extractant phase leaving E2 1544 flows into El mixer 1543 via line
1525. The aqueous phase exits E3 1546 via line 1529 and flows into the E4
mixer 1547 along with extractant phase from the storage tank 1597 which
flows via pump 1565 and line 1501. The extractant phase exits E3 1546 via
line 1505 and flows into E2D mixer 1545. The aqueous phase leaving E4
1547 flows to the 0/W separator 1598 via line 1502. The extractant phase
leaving E4 1547 flows to E3 1546 via line 1504. The 0/W effluent exits via
line 1503 as the purified water 1599. The extractant phase in the 0/W
separator 1598 exits via line 1535 and flows into S1-MS04 mixer 1541.
Referring now to Figure 17, this figure illustrates a presently preferred
configuration: M are mixers, El to E4 are extractor units with associated
decanters and weirs having flow control guides, and S1-504 to 54-SO4 are
the sulfate strippers. Typically, the extractor floc weirs have entrance and
exit ramps with a rounded lip and bottom. The configuration shows a water
purification circuit 1801m, a metal strip circuit 1803, and a sulfate strip
circuit
and extractant phase regeneration 1805. Metal recovery is in metal stripper
88
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
SIM where a metal sulfate concentrate product is obtained. Note that the
metal and sulfate strippers have internal recycle lines for aqueous recycle
flow.
The apparatus has an AMD feed water inlet 1800 and various
interconnections for flow referred to as aqueous lines 1853 and extractant
lines 1855. The AMD feed water 1800 flows via line 1802 into El mixer (M)
1832 along with extractant phase and floc from E2 decanter 1823. The
mixture flows past flow guide 1834 and over weir 1835 into El decanter
1833. In El decanter 1833, the floc thickens and maintains continuous flow
due to flow guide 1836 and floc weir 1835. The aqueous phase flows under
flow guides 1834 and 1836 and returns to E2 M 1822 via two standpipes and
line 1858 in El decanter 1833. The extractant phase and floc enters S1-M
mixer (M) 1840 along with the acidic aqueous phase via line 1856 from 52-M
1841. The floc is disintegrated by the acidic aqueous phase and forms acidic
aqueous and extractant phases. The aqueous phase leaving S1-M 1840 is the
metal sulfate concentrate product 1811 and can be collected via line 1808 in a
storage vessel. The extractant phase leaving S1-M 1840 flows into S2-M mixer
(M) 1841 via line 1818 along with the sulfuric acid solution 1813 which flows
via line 1810. The acidic aqueous phase exits S2-M 1841 via line 1856 and
flows into S1-M mixer (M) 1840. The extractant phase leaving S2-M 1841
flows into S1-SO4 mixer (M) 1842 with aqueous phase from S2-SO4 1843.
The aqueous phase exits S1-504 1842 as the sulfate concentrate product
1815 and is collected via line 1812. The extractant phase leaving S1-SO4
1842 flows into S2-SO4 mixer (M) 1843 with aqueous phase from S3-SO4
1844. The aqueous phase exits S2-SO4 1843 and flows into S1-SO4 mixer (M)
1842. The extractant phase exits S2-SO4 1843 and flows into S3-SO4 mixer
(M) 1844. The aqueous phase leaving S3-504 1844 flows into S2-SO4 mixer
(M) 1843. The extractant phase leaving S3-504 1844 flows into S4-504 mixer
(M) 1845 along with the carbonate solution 1817 which flows via line 1814.
The aqueous phase leaving S4-SO4 1845 flows into S3-SO4 mixer (M) 1844.
The extractant phase exits S4-SO4 via line 1816 as the regenerated
extractant solution 1819 and is collected in a storage vessel.
89
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
The aqueous phase withdrawn from El decanter 1833 via line 1858
and two standpipes flows into extractor E2 mixer (M) 1822. The extractant
phase in El decanter 1833 flows over flow weir 1835 and flows into Sl-M
1840. The aqueous phase flows through E2 1822 and exits via flow guides
1824 and 1857 and two standpipes to line 1859. The extractant phase and
floc leaving E2D 1823 flows into El mixer (M) 1832 along with AMD feed
water 1800 which flows via line 1802. The aqueous phase flows into E3 mixer
(M) 1821 via line 1859 with extractant phase from E4 1820. The aqueous
leaving E3 1821 flows into E4 mixer (M) 1820 via line 1860 with fresh
to extractant phase 1807 which flows via line 1804. The extractant phase
leaving E3 flows into extractor E2 mixer (M) 1822 via line 1861. The aqueous
phase flows through E4 1820 and exits via line 1806 as the purified water
product 1809. The extractant phase leaving E4 1820 flows into E3 mixer (M)
1821 via line 1862.
To demonstrate that the selected process of the invention was
scaleable, a field demonstrating unit was constructed with process flow
diagram similar to that of Figure 15 but sized to be operable at 5 to 40
gal/min, and optimal at 12 to 30 gal/min acid mine drainage water feed rate.
The field unit was the size of two tractor trailer beds and located on an
abandoned mine site at St. Michael, PA where a continuous acid mine
discharge water is flowing at a rate of 10,000 gallons/min so that sufficient
feed water was available for 24/7 operation. These trials lasted for 3 months
where the water purification process chemistry previously described was
confirmed at this larger scale. The E/A ratio was varied in this three months
of testing over the range of 1/ 2 to 1/10 and where the preferred ratio was
1/3 to 1/8, and most preferred ratio was the same as was found for the
laboratory and pilot testing, or 1/ 4 to 1/6. This testing demonstrated the
robustness of the technology, its good economics, ease of operation, and
water purification capability.
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
EXAMPLES
The following examples illustrate various aspects of the invention and
are not intended to limit the scope of the invention in any way.
Example 1
The objective of this test was to determine the key parameters and
ranges necessary for a successful removal of contaminants from acid mine
drainage water and accomplishing the separation of the recovered
components while forming them into concentrates. The process configuration
in Figure 13 was constructed using clear chemical resistant PVC (CPVC) for
the mixer-settler tanks, which had had an internal mixer volume of 186.7 cm3
and an internal settler volume of 401.5 cm3. The flow rate used resulted in a
333.6 second residence time. Clear Tygon tubing (0.25" I.D.) was used for
the piping. Cole-Parmer Instrument Company Master-flex L/S Peristaltic
pumps and Dayton AC-DC series motor mixers were used. The extractant
solution formulation was prepared using 15% Aliquat 134, 15% Exxon 10
(Isodecanol), and Calumet 400-500. All test conditions are given in the LLX
Test Condition Key.
The process was started up, operated, and shut down in the following
manner.
At start-up, the system was charged with aqueous solutions first, and
each mixer settler of the process was charged with approximately 50% of its
respective volume. The system is initially charged with 50% Sulfuric Acid,
followed by a 15% Sodium Carbonate solution. The more preferred method
is to charge the strippers with 25% H2SO4 to stay away from excessive strong
acid which can tend to third phase formation. Most preferred is to charge the
system with actual M-SO4 concentrate from a previous run. Charging the
system in this manner causes the extractant overflow receiving compartments
to partially fill. After this phase of start-up is complete the system is now
ready for the extractant solution.
The extractant solution must be fully acid stripped of Cl and then
carbonate loaded (1-25% Na2CO3), preferably 15% Na2CO3) before being
91
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
added to the process. Although the introduction of extractant solution is best
achieved at the pilot and commercial scale levels using pumps, at the
bench/lab scale this can be quickly achieved by manually pouring the
extractant solution into the mixer-settlers to the point of overflow into the
E-
phase overflow compartments. After charging the flotation liquid-liquid
extraction circuit with a sufficient volume of extractant solution, there
needs
to be enough extractant solution left in the surge tank so that the process
needs are met during normal operation. The total volume of the extractant
solution surge tank should be designed large enough so that it does not
.. overflow during the operation of the flotation liquid-liquid extraction
process
and can be charged with extractant solution when the system is shut down
between operations. The steady-state volume of the extractant solution in
the surge tank is then monitored visually or electronically with level
switches.
This should be done periodically so that the extractant solution surge tank
volume can be adjusted as needed to maintain sufficient extractant solution
volume to provide steady operation over extended periods, for example
weeks, months and possible years. The stirrers for the mixers were then
powered up, adjusted and maintained at steady-state by the following
procedures.
All of the mixers were set between 700-1700 rotations per minute
(rpm). The mixers need at least 15 minutes to warm up, preferably 30
minutes. During this time the mixers are monitored and adjusted, usually
decreasing the rotation rate in order to avoid excessive mixing. Excessive
mixing is very undesirable. It can lead to problems such as spatter as well as
the formation of fine emulsions that may be stable or that require longer
phase coalescence time in the settlers. Although any type of stirring is
sufficient enough to mix medium to low viscosity immiscible fluids, disk or
fin
type stirrer pumps are preferred. They are both designed to pull the two
fluids, aqueous and extractant solution, into the mixing compartment from
the upstream mixer settlers. The shearing blades of the mixers generate
micro droplets that create a very high interfacial surface area that is
critical to
fast contaminant extraction and strip kinetics. Higher mixing speeds
92
CA 02911119 2015-11-03
,
WO 2008/100610
PCT/US2008/002092
accommodate shorter residence time of the fluid in the mixer and compensate
for extractant/aqueous ratios other than 1:1. Although stirrer speeds that
result in the mutual blending of only 20% of the two phases is sufficient, a
blend of at least 80% is preferred and if optimum conditions can be achieved
95-100%. Excessive mixing is suitable but less preferred if the resultant
emulsion formed requires mixing for long periods of time to disengage and
break due to exceedingly fine droplet size. Mixing conditions preferred by the
invention is about 12-120 seconds, preferably 30-90 seconds, and if optimum
conditions can be achieved 45-seconds. The total hydraulic fluid residence
time in the mixer and the settler necessary for this process should be 10
times that amount or approximately 15 minutes. Due to the low values
and/or quantities of the contaminants present in the Acid Mine Drainage
water purification process (Fe, Al, Se, Si, Mn, Zn, Ni, Co, Ca, SO4), and the
very high flow rates of 10-10,000 gal/min (averaging about 500 gal/min, but
often variable) of water, conventional metals extraction by liquid-liquid
extraction is not feasible because of the very large equipment and E/A ratio
requirements that would be required for the large aqueous flows involved in
water purification. Although the mixing conditions can be either extractant
phase continuous or aqueous continuous, the latter is the more typical since
special startup conditions are not needed to achieve it.
After this initial loading of strip solutions and the warm up time for the
mixers are both complete, the system is now ready for the charging of the
acid mine drainage feed water and {the acid mine drainage feed pump was
set to 72 ml/min at system start-up (test parameter goal for acid mine
drainage feed stream is 100 ml/min)} the extractant phase feed streams.
The N-extractant (extraction circuit designed to extract +2 metal ions) feed
flow rate at system start-up is set to 18 ml/min. The extractant surge tank
for this process was a 4L clear chemical resistant PVC tank. Other feed
agents are listed in the LLX Test Condition Key.
Operation, Control, Monitoring (approaching and maintaining steady-
state): The process is run for approximately 20 hours before steady-state is
reached. This gives the extractant phase enough time to cycle through the
93
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
process at least 3 times (assuming 6L surge tank used @ 15 ml/min). The
extractant phase contacts the feed first during the metal extraction stage and
then gets metal stripped with Sulfuric Acid (500/0 H2SO4). After the Metal
Strip Stage the extractant gets carbonate (15% Na2CO3) loaded to extract the
sulfate ions and is now ready for use again. The extractant phases flow
scheme is also illustrated in Figure 13.
Once the extractant has had enough time to cycle through the system
at least once approximately 80% of the extractant phase stays within the
system and the remaining 20% stays in the surge tank. The acid mine
lo drainage feed rate was increased in 10 ml/min increments until the test
parameter of 100 ml/min was reached. Occasionally the system must be put
in idle mode (park) until certain control issues can be adjusted, any chemical
additions can be added, or any maintenance issue can be addressed. When
this is done all of the feed pumps are shut off and the mixers are allowed to
continue to circulate the process fluids. For example, for this test the
process
was parked for the following reasons: extractant feed levels running too low,
recalibration of pumps needed, aqueous in extractant feed pump, etc. Once
the process is said to have reached steady-state data readings can be
collected and sampling can now take place.
The samples for this test were collected out of the metal and sulfate
strip extraction stages as well as the acid mine drainage Feed and Raffinate
discharge lines (E1-E2-E3-E4-S1SO4-S2SO4-S3SO4-S4SO4-Feed-Raffinate).
There were two samples taken in 1 L bottles from the acid mine drainage
drum per run for analysis (sample with 2% Nitric Acid and "as is" sample).
Each extraction stage sample was taken from the aqueous phase of
the fluid in the settler tank using plastic disposable Luer Lock syringes, and
filtered using Serum Acrodisc 37 mm syringe filters with glass fiber (GF)/0.2
micron pores. This precautionary measure was taken to assure a minimal
amount of organic phase within the sample. The samples were taken within
one hour of each other. There were four samples taken from this run. The
data collection should be done in collaboration with the sample collection
every hour, it consists of: pH, density, E:A ratio, chemical volume,
extractant
94
CA 02911119 2015-11-03
,
WO 2008/100610
PCT/US2008/002092
=
depth, and mixer tip speeds. Once the data has been collected it will
immediately be place inside of the lab notebook. The samples are then sent
off for Inductive Coupled Plasma (ICP) and Inductive Coupled Plasma Mass
Spectrometry (ICP-MS) analysis.
At the end of this test 37.2 gallons of add mine drainage feed had
been used over the course of approximately 40 hours.
Example 1A (Stages determination)
This example determined the effect of sulfate extraction efficiency
using extractant phase formulation. The apparatus of Figure 13 was
constructed. The extractant phase was prepared by selecting components
from Table 3, and certain components of these were selected and blended in
proportions given in Table 5. The test conditions used are given in Table 5.
The experiment used extractant solution from Table 5 to extract sulfate
from 2000ppm Na2SO4 solution. Since the extraction of sulfate anions in a
Na2SO4 solution rely mainly on a concentration gradient, this set-up served as
"worst case" in sulfate anion removal of water purification process. The
extractant solution was treated with 15% Na2CO3 to load up the extractant
with carbonate anion, and then the extractant and Na2SO4 solution were
tested under different E/A ratio. The aqueous phase was analyzed using ion
chromatography to determine the remaining sulfate concentration. The ion
chromatography results determined the amount of sulfate removed at
different E/A ratio. And those results were also plotted via McCabe-Thiele
plot, which lead to the discovery of the stages needed for sufficient sulfate
loading and stripping of the water treatment process. The experiment also
determined the range of extractant needed for effective sulfate extraction,
which preferred by the invention is about 5-15%, most preferably 8-12%.
The McCabe-Thiele result is shown in Figures 9 to Figure 12.
To validate the results, a sulfate extraction process was set up with the
apparatus of Figure 13. This process was designed to extract sulfate anion
from a 2000ppm Na2SO4 solution via a continuous circuit. The result from
this process validated the proposed design and the same sulfate process
CA 02911119 2015-11-03
=
WO 2008/100610
PCT/US2008/002092
=
apparatus was implemented into the overall process flow scheme. This also
established a baseline test condition, which includes E/A ratio between 1/4-
1/6 for sufficient sulfate extraction and stripping in three to four stages.
Example 2 (Run #2)
This example determined the effect of metal and sulfate extraction
efficiency in a less contaminated stream using extractant phase formulation.
The apparatus of Figure 14 was constructed. The extractant phase was
prepared by selecting components from Table 3 blended in proportions giving
in Table 5. The test conditions used are given in Table 5.
This experiment used extractant solution from Table 5 with process
apparatus of Figure 14 to perform water purification process with an acid
mine drainage stream containing low contaminant levels. The process was
set up with the same number of extracting and stripping stages. The
combination of high extraction efficiency of metal cation and sulfate anion
and the low contaminant concentration in the acid mine drainage stream
caused secondary emulsion at the third extraction stage. The emulsion also
stopped phase disengagement between extractant solution and the raffinate.
The extractant solution and raffinate emulsion created from the process
behaved similarly to an emulsion of an extractant solution and deionized
water. The emulsified raffinate from this process was left in the tank for 24
hours to prolong the settling time in an attempt to achieve better phase
disengagement. However, there was no visible phase separation after 24+
hours of settling time, and the emulsion was not able to break until pH
adjustment was made by addition of sulfuric acid. It appears that mixing was
excessive thus forming a stable microemulsion. Stable in that the
disengagement from the emulsion was too slow. When mixer speed was
controlled this problem did not return.
Example 3 (Run #6)
This example determined the effect of acid mine drainage liquid-liquid
extraction (LLX) system efficiency with shorter mixing residence time using
96
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
extractant phase formulation. The apparatus of Figure 14 was constructed.
The extractant phase was prepared by selecting components from Table 3
blended in ranges given in Table 4. The test conditions used are given in
Table 5.
This experiment also used extractant solution from Table 5 with
process apparatus of Figure 14 to evaluate acid mine drainage water
purification process performance under shorter mixing residence time. The
two-part experiment tested different process conditions; the first part of the
experiment tested the treatment process with E/A ratio of 1/6 and mixing
io residence time of 60 seconds, and the second part tested the treatment
process with E/A ratio of 1/8 and mixing residence time of 35 seconds.
The 60 seconds mixing residence time trial was behaving well on its
own, and the consumption rates of Na2CO3 and H2504 were determined in
this experiment. The raffinate from individual stream was sent to ion
chromatography and Inductively Coupled Plasma Mass Spectrometry (ICP/MS)
to determine the final metal and sulfate concentration. These results and
measurements helped to determine the final mass/molar balance of the
overall process. The 35 seconds mixing residence time trial also had a
smooth operation. However, the raffinate's ion chromatography and ICP/MS
results from this trial turned out not fulfilling the performance requirement.
This allowed the determination of the mixing residence time and E/A ratio
limitation for the treatment system. The analytical results from these trials
are shown in Table 6.
The result from the two-part experiment not only showed the limitation
zs of E/A ratio and mixing residence time needed for sufficient process
treatment, but also determined the extractant solution input rate with respect
to the mixer size. If too much extractant solution pushed into strippers, the
mixer section would be overwhelmed and started plugging up because of the
excess extractant solution in the mixer. The excess extractant solution in the
mixer had gel-like behavior and caused the mixer to lose hydraulic suction.
This would severely cripple the process because it was essentially equal to
shutting down the process at midpoint while more acid mine drainage water
97
CA 02911119 2015-11-03
e
WO 2008/100610
PCT/US2008/002092
k
and extractant were being pushed through the front end of the process. The
discovery of this particular limitation allowed the determination of the
maximum extractant solution flow rate into the strippers, thus provided the
boundary condition needed for the process.
Example 4
This example determined the effect of water entrainment from
extractant phase formulation, especially the contribution of chosen extractant
and diluent and their relationship. The test apparatus consisted of a series
of
graduated, capped vials used to perform batch evaluations of the degree of
water entrainment with respect to extractant phase formulation at extraction
and strip conditions, and while preserving good yields of sulfate ion
extraction. The extractant phase was prepared by selecting components from
Table 3; blended in proportions given in Table 5. The test conditions used
is are given in Table 5.
This series of experiments used extractant solution from Table 5 to
determine the "best case" extractant phase formulation that yielded the least
water entrainment in the extractant phase. The experiments were designed
by using Design Expert, version 6.0, and the goal for this experiment was to
discover the extractant phase formulation range that would potentially
produce the highest metal sulfate product without compromising the sulfate
extraction efficiency. There were five different concentrations of extractant
solutions and each was conditioned with 15% Na2CO3. The different
extractant solutions were thoroughly mixed with acid mine drainage water
with E/A ratio of 1/6. The mixtures were then left to settle and the settling
time was recorded. A certain amount of extractant phase was transferred to
a centrifuge to pull any entrained raffinate out from the extractant phase.
The leftover raffinate was then transferred out and the extractant phase was
then treated with 50% H2SO4 for metal extraction. Measurements such as
phase disengagement time, amount of raffinate entrained, color of the
extractant phase, etc. were taken during the experiments and the results
were entered into the Design Expert software.
98
CA 02911119 2015-11-03
=
WO 2008/100610
PCT/US2008/002092
Figure 16, from Design Expert version 6.0, showed schematically the
relationship of the concentration of Aliquat and Isodecanol and their impact
on post-phase separation water entrainment in the extractant solution. The
goal was to find a "best case" formulation and test condition with respect to
s high metal sulfate production. This experiment not only led to the
discovery
of the "best case" test condition needed for producing high metal sulfate
product, but also demonstrated the effect of the rapidly declining metal
sulfate concentration caused by water entrainment. If the raffinate water
was entrained in extractant solution, even a small amount of water would
reduce the metal sulfate concentration dramatically and sacrificed the
salability of the metal sulfate product. The diluted metal sulfate
concentration
would also need a long time to build back up to a desirable level.
Example 5 (Run #7, Scheme 3)
This example determined the effect of the optimum extractant phase
formulation found in the previous designed experiments (example 4) and its
influence on metal sulfate product concentration. The apparatus of Figure 3
was constructed. The extractant phase was prepared by selecting
components from Table 3 and blended in proportions giving in Table 4. The
test conditions used are given in Table 5.
This experiment used extractant solution from Table 5 with process
apparatus of Figure 3 to evaluate the extractant phase "best case"
formulation with respect to amount of water entrainment in the extractant
phase and the concentration increase in the metal sulfate product stream.
The experiment was set up to test several different test conditions, and a set
of measurements were conducted from each of the test conditions to
determine the metal sulfate product flow rate and the consumption rate of
sulfuric acid. These measurements were critical for determining the metal
sulfate production rate and provide a more realistic economic estimation for
the field-scale process unit. The results from the experiment were then
entered into the Design Expert software, version 6.0 to determine the
optimum test condition for producing high concentration metal sulfate product
99
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
stream. Test condition range and the optimum test condition that would be
needed to produce high metal sulfate concentrations were determined. As
determined by the software, the E/A ratio would be increased from 1/6 to
1/4.35, with longer mixing residence time of 112.5 seconds. The result from
this experiment will serve as the test condition for the next experiment, and
the metal sulfate product concentration would be compared with the previous
tests and a final extractant phase formulation could be determined.
Example 6 (Run #8)
io This example determined the effect of the "best case" extractant phase
test condition found in the previous designed experiments (example 5) and its
influence on metal sulfate product concentration. The apparatus of Figure 3
was constructed. The extractant phase was prepared by selecting
components from Table 5 and blended in proportions giving in Table 4. The
test conditions used are given in Table 5.
With the previously determined test condition, the experiment ran over
27 hours without major problems and was also able to provide valuable
information on the metal sulfate product concentration. The flow rate of
metal sulfate product was determined, and the ratio of metal sulfate product
to acid mine drainage feed flow rate were about 1/5 or less.
The process also added sodium hydroxide solution into the last stage
of extraction in an attempt to extract out additional magnesium via pH
control. And some solid particles precipitated and settled in the bottom of
the
last extraction settler. This also indicated the potential product that could
be
produced from this process and also the additional implementation to avoid
solid blockage inside the transferring lines. The results for this experiment
are shown in Table 8
This experiment not only showed the potential concentration of the
metal sulfate product, but also the sensitivity of such product stream toward
water carry-over by the extractant solution. Even a small amount of
water/raffinate carried over by the extractant solution would decrease the
concentration tremendously. And once the concentration dropped, it would
100
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
take a long time to build up the concentration back to the desirable level.
The diluted metal sulfate product stream would also create faster harvesting
flow rate, i.e. the ratio of metal sulfate product to acid mine drainage feed
flow rate would increase to 1/3 or more. The sudden surge of water/raffinate
disrupted the level of the extractant and aqueous phase and caused domino
effect to all the downstream process. Therefore, it was essential to find a
good control method for metal sulfate stripper to guarantee the success of
this process.
io Example 7 (Run #9)
This example determined the effect of the extractant phase test
condition with alternative diluent in the formulation and its influence on
metal
sulfate product concentration. The apparatus of Figure 3 was constructed.
The extractant phase was prepared by selecting components from Table 5
and blending in proportions giving in Table 4. The test conditions used are
given in Table 5, .see Run #9 in this table where Scheme 3 denotes the
apparatus in Figure 3.
This experiment evaluated' the metal sulfate product concentration and
the presence of water/raffinate in the extractant solution with alternative
diluent. Additional bench-scale testing showed that incorporating aromatic
diluent in extractant phase formulation would yield low water/raffinate
entrainment in the extractant solution, thus lower the water/raffinate carry-
over into the metal sulfate strippers. The extractant phase formulation with
aromatic diluent behaved well in the same process configuration. And the
alternative extractant phase formulation performed as expected and
significantly decreased the water/raffinate carry-over into the metal sulfate
strippers.
The test condition of this experiment was kept at baseline process
condition, i.e. 60 seconds mixing residence time, E/A ratio = 1/6. Testing
with the baseline process condition provided grounds for comparison between
different extractant phase formulation and the effect of using different
diluent. The formulation with aromatic diluent showed the metal oxide
101
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
colloids in extractant solution were flowing well and did not create clumps of
metal oxide floc. This definitely helped the transfer efficiency of metal
oxide
colloids into metal sulfate strippers due to the fluidity of the solution.
Unfortunately, the process had some water carry-over during part of the
experiment, which decreased the metal sulfate concentration. However, once
the process was back to steady state condition, the metal sulfate
concentration would build up quickly.
This experiment provided grounds for baseline comparison between
extractant phase formulation effect with respect to aliphatic and aromatic
diluent. This experiment had demonstrated the efficiency of using aromatic
diluent for extractant phase formulation. However, aromatic diluent had long
term material compatibility problem with the current setup, which provided
the basis for determining an optimum extractant phase formulation with
proper diluent mix. The optimum extractant phase formulation would have
low water/raffinate entrainment in extractant phase while being compatible
with easily accessed materials, such as PVC or fiberglass reinforced plastics.
Table 4. Extractant phase formulation table with typical minimum and
maximum extractant component. The extractant, modifier, and diluent can
be referred to Table 3.
Extractant phase Formulation
Extractant Volume Percentage
Formulation (%)
Component
Minimum Maximum
Extractant 5 15
Modifier 2.3 15
Diluent 70 92.7
In a broader aspect of the invention the formulations may comprise the
following (in volume %):
Extractant: 0.5 minimum to 70 maximum;
Modifier: 0 minimum to 95.5 maximum;
Diluent: 0 minimum to 95.5 maximum.
102
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
The important criteria that is required within the narrow or broader
material limits is that a floc is formed that separates from the treated
aqueous
phase and the loaded extractant phase.
Example 8
This example illustrates the determination of oil water separator and
extractor setting time and the determination of the acid and base
consumption rate during purification of acid mine drainage water using the
invention.
The continuous process configuration in Figure 9 was constructed
using clear, chemical resistant PVC for the mixer-settler tanks, which had an
internal mixer volume of 180 ml. This allowed the process fluids to have a
residence time of 60 seconds with the ETA total (combined) flow rate of
180m1/min. Additional equipment included, clear Tygon tubing for the piping
(0.375" I.D.), Cole-Parmer Instrument Company Master-flex US Peristaltic
pumps, and Dayton AC-DC series motor mixers. The extractant phase
formulation was prepared using 15% (v/v) Aliquat 1348, 15% (v/v) E>o<al 108
(Isodecanol), and 70% (v/v) Calumet 400-500 diluent. This diluent is less
than 1 wt% aromatics. Other test conditions are given in Table 5.
Relative to example 4, the process alterations made were:
1. The M & N extraction box decanters were replaced with separatory
funnels to allow fluid dimensions and sharp phase separator control.
2. The M & N Sulfate product discharge lines fed directly into separate
tanks.
3. The M Sulfate decanter was modified by drilling angled holes into the
mixer overflow weirs to facilitate a discharge of equal or lesser height of
the
El M Sulfate extractant phase flow.
4. A peristaltic pump was installed to transfer the M, N metal ions
(trivalent and divalent) and SO4 loaded extractant phase(s) to the appropriate
stripper.
103
CA 02911119 2015-11-03
WO 2008/100610 PC
T/US2008/002092
5. The extractant phase to aqueous acid mine drainage /extractant phase
ratio was changed to 1:5.
6. An oil/water separator was added to recover any extractant phase lost
from the extraction operation to the aqueous raffinate exit stream of the E4
extraction stage.
7. Double wide settler used for E3 and E4 (Figure 3).
8. To determine the impact of settling time during the extraction
operation.
The process was started up, operated, and shut down in the following
manner:
Chemical Charging (Start-up): The system was charged with aqueous
solutions, with each mixer settler of the process charged with approximately
50% of its respective volume. The strippers were initially charged with 5-
50% (w/w) sulfuric acid. The extractant phase regeneration mixer-settlers
were charged with a 15% (w/w) sodium carbonate solution. By charging the
system in this manner, typically the extractant overflow compartments fill to
half full capacity. After this phase of start-up is complete the system was
now ready for the extractant phase.
Fresh extractant phase was optimally pre-cleaned prior to first charging
to the extraction system by fully acid stripped (0.1 50% v/v H2SO4,
preferable 25%) and then carbonate loaded (15% w/w Na2CO3 (range 0.5-
30%) before being added to the process. Although the introduction of
extractant phase is best achieved at the pilot and commercial scale levels
using pumps, at the bench/lab scale this can be quickly achieved by pouring
the extractant phase into the mixer-settlers to the point of overflow into the
settler compartments. After charging the liquid-liquid extraction (LLX)
circuit
with a sufficient volume of extractant phase, there should be enough
extractant phase left in the surge tank so that the process needs are met
during normal operation. The total volume of the extractant phase surge tank
should be large enough so that it does not overflow during the operation of
the LLX process and can be charged with extractant phase when the system
is shut down between operations. The steady-state volume of the extractant
104
CA 02911119 2015-11-03
s
., WO 2008/100610
PCT/US2008/002092
phase in the surge tank is then monitored visually or electronically with
level
switches. This should be done periodically so that the extractant phase surge
tank volume can be adjusted as needed to maintain sufficient extractant
phase volume to provide steady operation over extended periods, for example
weeks, months and possibly years. The stirrers for the mixers were then
powered up, adjusted and maintained at steady-state by the following
procedures.
All of the mixers were set between 700-1700 rotations per minute
(rpm). The mixers needed at least 15 minutes to warm up, preferably 30
minutes. During this time the mixers were monitored and adjusted, usually
decreasing the rotation rate in order to avoid excessive mixing. Excessive
mixing is very undesirable; it can lead to problems such as spatter as well as
the formation of fine emulsions that require longer phase coalescence time in
the settlers. Although any type of stirring is sufficient enough to mix medium
to low viscosity immiscible fluids, disk or fin type stirrer pumps are
preferred.
They are both designed to pull the two fluids, aqueous and extractant phase,
into the mixing compartment from the upstream mixer settlers. The shearing
blades of the mixers generate micro droplets that create a very high
interfacial surface area that is critical to fast contaminant extraction and
strip
kinetics. Higher mixing speeds accommodate shorter residence time of the
fluid in the mixer and compensate for extractant/aqueous ratios other than
1:1. Although stirrer speeds that result in the mutual blending of only 20% of
the two phases is sufficient, a blend of at least 80% is preferred and if
optimum conditions can be achieved 95-100%. Excessive mixing is suitable
but less preferred if the resultant emulsion formed requires mixing for long
periods of time to disengage and break due to exceedingly fine droplet size.
Mixing conditions preferred by the invention is about 12-120 seconds,
preferably 30-90 seconds, and optimally 45-seconds. The total hydraulic fluid
residence time in the apparatus will be the sum total of the volumes of the
individual operations of the apparatus, including mixers, settlers, pumps and
surge capacity. Due to the low values and/or quantities of the contaminants
present in the Acid Mine Drainage water purification process (Fe, Al, Se, Si,
105
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Mn, Zn, Ni, Co, Ca, SO4-2), and the very high flow rates of 10-10,000 gal/min
(averaging about 500 gal/min, but often variable ) of water, conventional
metals extraction by LLX is not feasible because of the very large equipment
and E/A ratio requirements that would be required for the large aqueous
flows involved in water purification. Although the mixing conditions can be
either extractant phase continuous or aqueous continuous, the latter is the
more typical since special startup conditions are not needed to achieve it.
After this initial loading of strip solutions and the warm up time for the
mixers are both complete, the system was now ready for the charging of the
.. Acid Mine Drainage feed water and (the acid mine drainage feed pump was
set to 145 ml/min at system start-up) the extractant phase feed streams.
The N-extractant (extraction circuit designed to extract +2 metal ions) feed
flow rate at system start-up was set to 28 ml/min, and the M-extractant
(extraction circuit designed to extract + 3 metal ions) feed flow was set at
7.3
ml/min. The extractant surge tank for this process was a 6 L clear chemical
resistant PVC tank. Other feed agents are listed in the LLX Test Condition
Key in Table 5.
106
=
=
,
4
Table 5. LLX Test Condition Key
o
,,..
c.,
(extractant phase test conditions with extractant formulation selection for
each test trial.) c>
ot
.
=
=
e)
LLX Test Condition Key
5
a
Extractant Flow Extractant Flow I Mixing
AMD Flow Rate Rate (N Circuit) Rate (M Circuit) Residence Time
Extractanl Process
Run X mUmin mUmin mlimin EtA Ratio second
Formulation Test Objective Configuration Note
P
AMD Flow Rate Rate tN Circuit) Rate (M Circuit) residence Time
ExtractaniAliquat 134: 15%
1 100 17 8 116 333-6 (6443n1L -IDA: 15% .. Basic (IX
process evaluation
Scheme 1
mixer-settler) Calum 70% and key parameter range
finding
et:
0
Aliquat 134: 15%
333.6 (640m1
Added ceramic milling rods in o
3 100 17 a 1/6 IDA: 15% LLX process evaluation
Scheme 1 tv
mixer-settler) 0/W separator ic)
Calumet: 70%
Aliquat 134: 15% tlY process evaluation and
' Added ceramic muting rods in
307.2 (640mL
1-.
4 100 20 5 1/5 IDA: 15% Determining 15% Na2CO3
Scheme 1 01W separator. Used sep. i-.
mixer-settler)
l0
Calumet 70% consumption rate _
funnels as decanters
Afiguat 134: 15% LLX process evaluation and
tv
5.1 100 20 5 1/5 90 (180mt mix 'IDA: 15% Determining
15% Na2CO3 Scheme 1
o
settler)
Added air floatation device and i-.
_______________________________________ Calumet: 70% consumption rate
ceramic milling rods in OM/ 1
60 (180mL mixer Aliguat 134: 15% LLX process evaluation and
5.2 145 28 7.3 1/5 settler) 10k 15% Determining
acid and base Scheme 1 separator i-.
1
Calumet: 70% consumption rate
0
u.)
60 (180mL mixer
Niguel 134: 15% LLX process evaluation and
IDA: 15%
6.1 154 26 N/A 1/6 Determining he acid
and base Scheme 2 Run E4 extractant FR at
settler)
_______________________________________ Calumet 70%
consumption rate 400/min (0-1=10). Added
35 (180m1 mixer'Aliquat 134: 15% flotation 0/F attachment
LLX process evaluation and key
6.2 275 35 NIA 118 IDA: 15% Scheme 2
Extractant inlet: El, 52, and E4.
settler) parameter range finding
_ . Calumet: 70%
- - -.... _
' Aliquot 134:91% Statistically designed test
to _ Changed El and E2 to T-
7 - - - - - IDA: 4.3% determine "best case"
testing Scheme 3 stepped mixer-settler (Scheme
Calumet: 86.6% _condition
3)
' Aliquat 134: 9.1%
Test condition determined by rl
LLX process evaluation and key
a 78.1 17.9 NIA 1/4.35 112.5 IDA: 4.3%
Scheme 3 Run X7 based on maximizing 2
parameter range finding
IMS041 product.
Calumet: 86.6%
on
-
la
Aliquat 134:9.1% LLX process evaluation with
0
o
9 154 26 N/A 1/6 60 IDA: 4.3% Aromatic
diluent and the impact Scheme 3 Extractant inlet: El, E2, and E4 co
Aromatic 150: 86.6% of IMS04) product
=
o
...
i,.=
o
\,e
t..)
107
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Operation, Control, Monitoring (approaching and maintaining steady-state):
The process was run for approximately 20 hours to insure steady-state
concentration was reached. This amount of time gives the extractant phase
enough time to cycle through the process hardware at least 3 times (6 L of
extractant phase with surge tank and regenerates the extractant phase
leaving it 15 ml/min). The extractant phase contacts the feed first during the
metal and sulfate ion extraction stage and then the metals are stripped with
sulfuric acid (initially 50%v/v H2SO4). After the metal ion strip stage the
extractant was regenerated using counter-current flow with 15%w/w Na2CO3
to strip the sulfate ions and regenerates the extractant phase leaving it
ready
for use again. The extractant phases flow scheme is illustrated in Figure 10.
Once the extractant has had enough time to cycle through the system,
at least once, approximately 80% of the extractant phase stays within the
system and the remaining 20% stays in the surge tank. The fact that the
apparatus can be turned off and on quickly is an important operational
advantage. Occasionally the system was put in "idle" mode (parked) until
certain controls or hardware could be made or addressed chemical additions
could be added, or any maintenance issue could be addressed. When this
was done all of the feed pumps were shut off and the mixers were allowed to
zo continue to circulate the process fluids in the mixer-settler. For
example, for
this test the process was parked for the following reasons: extractant phase
in E1D was too thin. The E1D decanter, with the drilled holes, allowed the
aqueous phase at a pH of 3.35 to enter the SIM stripper, the SIM and S2M
mixer settlers became aqueous flooded causing the combined extractant and
aqueous phases from S2M to enter S1-SO4 causing it to contain too much
aqueous phase. Once the process reaches steady-state, data readings were
collected and samples were taken long enough without upset of steady-state
conditions to achieve steady-state (three (3) turnovers of extractant phase in
the system).
Sampling and Data Collection: The samples for this test were collected
from the metal ion and sulfate ion strip and extraction settlers, as well as
108
CA 02911119 2015-11-03
, WO 2008/100610
PCT/US2008/002092
from the acid mine drainage feed and raffinate discharge lines (El, E2, E3,
E4, S1M, S2M. S1N, S2N, S1SO4, S2SO4, S3SO4, S4SO4, Feed-Raffinate).
Only aqueous phases were sampled.
109
,
,
o
Table 6. Analytical result from Run #6.1
,...A
G,
c,
with 60 seconds mixing residence time and E/A ratio of 1/6. co
,...
c.
Run #6.1: 60 Second Mixing, E/A r- 1/6 .
o
Sample Position. Sulfate
Magnesium Aluminum Silicon Calcium Iron (total) Manganese, Cobalt
Nickle Zinc
Sample Unit ppm PPm PPm PINT1 , PPm PPm
PPm PPm PPm , PPm
AMD Stream 2512 142.1 1.204 5.623 152.2
234.0 78.77 0.9494 0.8324 1.633
Std. Dev. 1180 2.6 1.311 0.207 3.6
9.0 5.16 0.0316 0.0229 0.315
Concentrated
Sulfate Product
Stream 209982 39.55 14.25
10.25 81.35 495.6 99.82 1.931 1.584 8.138 o
5),
Std. Dev. 121274 3.91 8.18 6.73 55.40 853.4
100.63 2.454 2.017 4.179 0
Concentration
Na
l0
Average Data Factor , > 80x N/A N/A N/A N/A N/A
N/A N/A N/A N/A H
H
Metal (II) Sulfate
H
H
Product Stream 58728 136.9 12.07 12.13 178.7 693.5
162.1 2.922 2.420 2.499 l0
Std. 0ev. 27799 4.0 11.19 8.75 37.2 775.9
110.7 2.461 2.031 0.221 N.)
0
Concentration -
H
01
I
Factor NIA N/A > 10x > 2x > lx > 2x
2x > 3x > 2x > 1.5x r
Purified Stream _ 41.63 , 134.5 <0.1 <2.5 <10 <0.25
8.936 <0.01 <0.01 N/A
1
Std. Dev. 17.55 4.8 1.032 0.491 14.55
N/A 4.596 0.0043, 0.0571 N/A 0
_
w
Minimum
Target Achievement _ <500 <120 <1 -- <300 <1 <1 --
- -
Performance Drinking Water
Acheivement Standard < 250 < 80 < 0.2 - <150 <0.3
<0.3 -- - --
_
in Purified Phase 2
ot
n
Stream Screening
0-3
Objective <125 _<40 _<0.1 --
<125 <0.15 <0.15 - - --
uz
k4
o
o
oo
8
o
t.)
0
0
t4
1 1 0
,.
,
o
Table 7. Results from Run #6.2 with 35 seconds mixing residence time and E/A
ratio of 1/8 1,4
4=
=4,
co
Run #6.2: 35 Seconds Mixing, E.IA = 118
=
=
Sample Position Sulfate Magnesium Aluminum Silicon Calcium Iron (total)
Manganese Cobalt Nickle Zinc *OfG Analysis c,
..
.
o
Semple Unit , ppm ppm ppm ppm ppm ppm ppm ppm ppm
ppm mg/t.
AMD Stream , 1713 155.3 0.7332 5.661 151.7
239.9 84.12 0.957 0.8159, 1.444 NIA
-Std. 0ev. 105 6.5 0.5024 0.243 1.1 12.7
2.52 0.044 0.0309 0.133 NIA
Concentrated
Sulfate Product
Stream 236571, 36.64 4.833 11.10 70.02,
599.8 57.70 1.054 0.9989 2.218 N/A o
>
Std. 0ev. 64000 16.88 2.928 2.56 36.88
257.6 42.44 0.741 0.5827 1.246 N/A 0
_
N.)
Concentration
lt,
Average Data Factor > 130x N/A N/A WA N/A WA N/A
N/A N/A N/A WA r
1-,
Metal (II) Sulfate
F.
I-'
Product Stream 77392 184.3 18.49 18.31 327.0,
1388 301.1 4.932 4.085 8.974 NIA kr>
Std. 0ev. 58097 _ 25.8 12.49 8.52 120.5
863 141.5 2.466 1.991 5.671 WA N
0
Concentration
H
Lil
I
Factor , > 55x N/A > 25x . > 3x > 2x > 5x > 3x
> 5x > 5x > 6x NIA
r
Purified Stream 1605 136.3 <Di 3.811 70.73
126.5 19.48 0.2642 0.2392 <1 9.5 1-,
1
...
Std. Dev. 1201 19.4.. NIA 0.784 38.56
56.5 28.31 0.4044 0.3455 N/A N/A 0
u.)
Minimum
Target Achievement <500 <120 <1 - <300 <1 <1 -, -
- -
Performance Drinking Water
Acheivement Standard <250 _<80
_<0.2 - <150 _<0.3
_, <0.3 - - - -
in Purified Phase 2
Stream Screening
Objective <125 _<40
_<0.1 - <125 <0.15
_<0.15 - - - 10.00
ot
en
,..-i
*01G analysis refers to oil/grease analysis
tt
1
-6--
0
I.)
=
rsi
1.11
"
..
Table 8: Run #8 Results with 112.5 seconds mixing residence time and E/A ratio
of
1:4.35 0
t.)
Run #8: 112.5 Second Mixing, ElA = 1/4.35
=
c
Sample Position Sulfate Magnesium Aluminum
Silicon Calcium Iron (total) Manganese Cobalt Nickle Zinc 'VG
Analysis!!
Sample Unit ppm ppm ppm ppm ppm ppm ppm
ppm ppm ppm mg/L g
AMD Stream 1665 171.8 2.298 4.645 133.1
199.9 87.12 0.9517 0.8471 1.579 N/A ,?-='
o
Std. Dev. 87.77 1.9 0.245 0.102 3.7 6.1
0.71 0.0067 0.0149 0.162 NIA
Concentrated
Sulfate Product
Stream 131976 22.72 2.036 7.846 33.77 56.24 26.23
0.3500 0.3309 0.8110 N/A
Std. Dev. 12403 16.52 2.028 2.081 27.63
65.11, 35.68 0.4150 0.3459 0.5477 N/A
Concentration
Factor > 80x NIA N/A NIA N/A N/A N/A N/A NIA
N/A N/A r)
5=, Average Data
0
Metal (II) Sulfate
N.)
l0
Product Stream 256314 193.2 21.17 20.65 423.7
2174 693.5 8.236 7.258 13.139 N/A
I-,
Std. Dev. 182848 24.6 16.19 11.87 164.0
1415 446.7 5.689 5.106 8.923 N/A
I-.
Concentration
lt)
Factor N/A N/A >5x >2x > lx >3x >1.5x >3x
>2x >1.5x N/A .. N.)
0
Purified Stream 80.71 148.8 . <0.1 <2.5 <10
<0.25 0.0800 <0.01 <0.01 0.0741 27.6
cri
I
Std. Dev. 74.93 8.2 WA N/A N/A N/A
0.0761 N/A N/A 0.0338 N/A Minimum
Target
i
Target Achievement <500 <120 <1 -- <300 <1 <1
- -- -- - 0
w
Performance Drinking Water
Acheivement Standard <250 _<80 _<0.2 -- <150 _<0.3
<0.3 ' - -- - -
in Purified Phase 2
Stream Screening
Objective <125 _<40 _<0.1 --_ <125 <0.15
<0.15 -- - -- 10.00
Run #8 Test Condition:
iv
rl
E/A in El, E2E3, E4 =1/4.35
Mixing residence time = 112.5 seconds
c/)
Extractant formula: 9.1% Aliquat 134,4.3% Exxal 10, 86.6% Calumet 400-500
N
CD
0
GO
CD "0/G analysis result is from Lancaster Laboratory
N
0
µe,
N
112
Table 9. Results for Run #9 with 60 seconds mixing residence time and E/A
ratio of 1/6.
Run #9 used an aromatic diluent instead of aliphatic.
o
,...
=
=
co
,-
Run #9: 60 Second MIxInE, E/A =1/6
o
o
Sample Position Sulfate Magnesium Aluminum
Silicon Calcium Iron (total) Manganese Cobal Nickle Zinc '0/G
Analysis o
=-,
Sample Unit ppm ppm ppm ppm ppm ppm ppm t PPm
ppm ppm = mg/I. o
AMD Stream 1535 161.1 2.424 6.275 133.4 193.9
84.89 0.9609 0.8491 1.446 N/A
Std. Dev. sa 2.5 0492 0.268 2.0 2.2 1.66
0.0161 0.0168 0.033 N/A
Concentrated
Sulfate Product
Stream 155455
9.572 0.1947 8.571 14.26 17.08, 3.122 0.0366 0.0653 0.6134 N/A
Std. Dev. 9426 0.881 0.0128 0.337 1.30 3.26
0.589 0.0018 0.0115 0.0701 N/A
Concentration
Factor >100x N/A N/A N/A N/A N/A N/A N/A N/A
N/A N/A
Average Data
>
Metal (11) Sulfate
o
Product Stream 217119, 131.0 14.42 24.27 224.1 1541
404.3 5.994 5.114 9.370 N/A N.)
l0
Std. Dev. 81856 73 5.37 7.01 40.1 547 143.1
2.284 1.965 3.435 N/A i-.
Concentration
1-`
Factor N/A N/A > 5.5x > 3x > 1x > 7.5x > 4.5x '6x
> 6x > 6x N/A I-.
lt)
Purified Stream 19.48 136.7 <0.1 <2.5 <10 <0.25
0.489 <0.01 <0.01 0.0467 133.0
Std. Dev. 15.05 0.8 N/A N/A N/A N/A 0.279
N/A N/A 0.0530 N/A N.)
o
Minimum
FA
Target Achievement <500 <120 <1 - <300
<1 <1 - - - - 01
1
Performance Drinking Water
i-.
Acheivement Standard <250 _<80 <0.2 - <150 <0.3 <0.3 -
- - -
i
In Purified Phase 2
o
w
Stream Screening
Objective <125 <40 <0.1 - <125 <0.15 <0.15
- - - 10.00
9 Test Condition:
E/A in El, E2, E3, E4 = 1/6
Mixing residence time = 60 seconds
Extractant formula: 9.1% Aliquat 134, 4.3% Exxal 10, 86.6% Aromatic 150
ti
el
*0/G analysis result is from Lancaster Laboratory
I-4
cn
14
0
0
00
---
0
0
k=J
0
,Co
kJ
113
CA 02911119 2015-11-03
= WO
2008/100610 PCT/US2008/002092
Example 9
This example illustrates potassium sulfate (K2SO4) product production.
The sulfate concentrate was collected from the S1-504 exit stream of the
operating unit (see Figure 17) to purify acid mine drainage water (see Table
2B and 2C) and to determine whether K2SO4 solid might be prepared without
formation of system damaging K2SO4 crystals, which are very hard and
adherent. Initially, a test was conducted to determine the details of the
K2SO4 isolation process. For this test, a graduated cylinder was weighed and
used to collect approximately 300 mL of the sulfate concentrate. The
concentrate that was collected was produced from the low Al acid mine
drainage water (Table 2C). The weight and volume of the sulfate concentrate
was recorded and used to calculate the density of the concentrate. A 20 mL
sulfate concentrate sample was collected for carbonate and sulfate analysis to
determine the initial carbonate and sulfate concentrations. To achieve K2SO4
crystallization, K2CO3(s) was added to the concentrate. Since it is desired to
have a recycle solution that is 5.5 wt% K2CO3 and 6.5 wt% K2SO4 to enable
generation of solution that could be recycled directly back to stage 52-SO4
unit of the sulfate strip circuit without further treatment, approximately
82.5 g
K2CO3(s) was added to the sulfate concentrate in five 16.5 g increments. The
solution was mixed using a stir bar and the mixing time, settling time, color,
and temperature observations were recorded. It was observed that
crystallization of a white solid occurs immediately after K2CO3(s) addition
and
the increase in temperature is barely noticeable. Once the crystals settled,
gravity filtration was employed to capture the fine white crystals. A 5 p
filter
bag was used to filter the solution and the filtrate was collected in a
beaker.
It was noted that the solution filtered quickly through the bag and that the
filtrate was a clear, yellow solution. The yellow color is due to trace Fe
contamination and is of no consequence. The weight and volume of the
filtrate was recorded and used to calculate the density of the filtrate. A 20
mL
filtrate sample was collected for carbonate and sulfate analysis to determine
the carbonate and sulfate concentrations after K2CO3(s) addition and
114
CA 02911119 2015-11-03
. WO 2008/100610 PC
T/US2008/002092
filtration. The wet, ivory-colored solid was scraped out of the filter bag,
weighed, and collected in a small vial. For the 300 mL sulfate concentrate
collected, 1.05 g of ivory-colored solid K2SO4 was produced.
Example 10
The production process of Example 9 was scaled up 10-fold to
investigate the K2SO4 yield with excess K2CO3(s). For this scale-up process, a
4 L beaker was weighed and used to collect approximately 3 L of the sulfate
concentrate from the F-LLX operation. The weight and volume of the
concentrate was recorded and used to determine the density of the sulfate
concentrate. A 20 mL sulfate concentrate sample was collected for carbonate
and sulfate analysis to determine the initial carbonate and sulfate
concentrations. To achieve K2SO4 crystallization, approximately 825.0 g
K2CO3(s) was added to the sulfate concentrate in five 165.0 g increments.
The solution was mixed using a stir bar and the mixing time, settling time,
color, and temperature observations were recorded. It was observed that
crystallization of a white solid occurs immediately after K2CO3(s) addition
and
the increase in temperature is barely noticeable. Once the crystals settled,
vacuum filtration was employed to isolate the white crystals. A Buchner
funnel, filter paper, and pump were used to filter the solution and the
filtrate
was collected in a 4 L flask. It was noted that the solution filtered quickly
and
that the filtrate was a clear, yellow solution. The weight and volume of the
filtrate was recorded and used to calculate the density of the filtrate. A 20
mL
filtrate sample was collected for carbonate and sulfate analysis to determine
the carbonate and sulfate concentrations after K2CO3(s) addition and
filtration. The wet, ivory-colored solid was allowed to air dry for a few
hours,
scraped off the filter paper, weighed, and collected in a sample jar. A 2.0 g
non-hygroscopic solid sample was collected to determine the moisture
content of the solid. When running the low aluminum AMD water (see Table
2B), for every 3 L of sulfate concentrate processed, approximately 66.78 g
K2SO4 is produced.
115
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Additional gravity filtration tests were conducted to determine which
filter bag pore size would work best. For these tests, the following pore
sizes
were evaluated: 1pm, 5pm, and 25pm. In addition, the amount of K2CO3(s)
was varied to determine whether the same yield could be achieved using less
.. K2CO3(s). In the first round of testing, a 4 L beaker was weighed and used
to
collect approximately 3 L of the sulfate concentrate from the F-LLX pilot
unit.
The concentrate that was collected was produced from the high Al acid mine
drainage water (Table 2C). The weight and volume of the concentrate was
recorded and used to determine the density of the sulfate concentrate. A 20
mL sulfate concentrate sample was collected for carbonate and sulfate
analysis to determine the initial carbonate and sulfate concentrations.
Approximately 90.0 g of K2CO3(s) was added to the sulfate concentrate and
the solution was mixed using a stir bar. The 3 L solution was divided into (3)
1 L portions and each 1 L portion went through a separate filter bag. A
peristaltic pump was used to pump the 1 L solution into each filter bag. For
the 1pm filtration, the 1 L solution was divided into two segments: 420 mL
and 580 mL. The 420 mL solution was pumped to the filter bag at 70 mL/min
and the remaining 580 mL was pumped at 580 mL/min. For the 5pm
filtration, the 1 L solution was divided into two 500 mL portions and each
.. portion was pumped into the filter bag at 500 mL/min. For the 25pm
filtration, the 1 L solution was pumped at 1000 mL/min. The pumping times
for each filtration were recorded along with draining times and observations
about the filtrate and filtration process. For each filtration, a 20 mL
filtrate
sample was collected for carbonate and sulfate analysis to determine the
.. carbonate and sulfate concentrations. If any solids were present, the wet,
tan-colored solid was scraped out of the filter bag, weighed, and collected in
a jar. The results of each filtration are displayed in Table 10 and the
analytical results are displayed in Tables 11A and 1113.
116
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Table 10. Gravity Filtration Data
Filter Amount Amount
Concentrate Filtration
pore of Filtrate Observations Solid
Volume Time
size K2CO3(s) Collected
14 min 18 Colorless solution, small
1pm 90.0 g 1 L sec amount of white
solids 7.69 g
Light orange solution,
small amount of white
5pm 90.0 g 1 L 23 min solids 9.39 g
Cloudy orange solution,
large amount of white
251im 90.0 g 1 L
min solids 0.00 g
Table 11A. Carbonate Analysis
5
Sample Equivalents Equivalents Sample
% K2CO3
Sample ID/ Volume Initial pH
to pH 8.3 to pH 4.5 Density
(Porosity) of sample (w/w)
(mL) (eq/L) (eq/L) _(g/mL)
K2SO4 Conc. 2.000 3.53 x x 1.0853
Filtrate (1pm)
0.400 10.62 0.19 0.45 1.1075 3.83
Filtrate (5pm)
0.400 10.75 0.19 0.43 1.1018 3.65
Filtrate
(25pm)
0.400 10.59 0.20 0.44 1.1058 3.67
Table 11B. Sulfate Analysis
Sample ID/ Sample
Dilution Volume Observed
Calculated mg/L Actual mg/L mol/L of
(Porosity) (pL) mg/L K2SO4
K2SO4 Conc. 1000 _ 50.0 49.5 51.1 51100 0.293
Filtrate (1pm) 1000 _ 50.0 44.5 45.8 45800 0.263
Filtrate (5pm) 1000 50.0 44 45.2 44000 0.252
Filtrate (25pm)
1000 50.0 43.7 44.9 43700 0.251
Based upon the experiment data, it is clear that the 1p and 5p filter
bags would perform well in the field. In the second round of testing, the 1p
filter bag was used and the amount K2CO3(s) was increased from 90.0 g per 3
L of sulfate concentrate to 180.0 g per 3 L of sulfate concentrate to increase
the yield of solid K2SO4. In addition, the sulfate concentrate was pumped to
is the filter bag in three 3L additions at 1000 mL/min. The first two 3L
solutions
were pumped directly to the filter bag while mixing and the last 3L solution
was mixed and allowed to settle before being pumped to the filter bag. The
117
CA 02911119 2015-11-03
= WO 2008/100610
PCT/US2008/002092
pumping times for each filtration were recorded along with draining times and
observations about the filtrate and filtration process. For each filtration, a
20
mL filtrate sample was collected for carbonate and sulfate analyses. If any
solids were present, the wet, tan-colored solid was scraped out of the filter
s bag, weighed, and collected in a jar. A 2.0 g solid sample was collected
to
determine the moisture content of the solid. The results of each filtration
are
displayed in Table 12 and the analytical results are displayed in Tables 13a
and 13b below. In Table 14, the moisture content results are displayed.
Table 12. Gravity Filtration Results-1p Filter bag
Amount Total Amount
Concentrate
of Filtration Time Filtrate
Observations of Solid
Volume
K2CO3(s) Collected
Colorless solution, very few
3 L 180.0 g 32 min 38 sec white solids
Colorless solution, very few
3 L 180.0 g 24 min white solids
Colorless solution, very few 284.43 g
white solids (per 9 L
3 L 180.0 g 25 min 20 sec
concentrate)
Table 13A. Carbonate Analysis-lp Filter bag
Sample ID Sample Initial Equivalents
Sample
Equivalents to %
K2CO3
Concentrate/ Volume pH of to 8.3 Density
P 4.5 'e 'L'
(w/w)
Volume (mL) sample (eq/L) g/mL
Filtrate 1t/3 L 0.400 10.85 0.41 0.88 1.1075
7.19
Filtrate 2""1/3 L 0.400 10.86 0.41 0.86 1.1100
6.94
Filtrate 3rd/3 L 0.400 10.86 0.45 0.95 1.1161
7.71
Supernatant
r/3 L
0.400 10.84 0,41 0.88 1.1134 7.23
Table 13B. Sulfate Analysis-1p Filter bag
Sample ID/
Dilution Observed mg/L Calculated mg/L Actual mg/L mol/L of K2SO4
Volume
Filtrate 1st 3 L
1000 37.1 49.2 49200 0.282
Filtrate 2nd 3 L 1000 37.3 49.4 49400 0.283
Filtrate 3rd 3 L 1000 36.9 48.9 48900 0.281
Supernatant
VI 3 L 1000 38.0 50.3 50300 0.289
118
CA 02911119 2015-11-03
WO 2008/100610
PCT/US2008/002092
Table 14. Moisture Content Results
Initial Final Weight Final Wt.
Sample Tare Weight Initial Solid 0/0
Weight (g) (9) (9)
ID (9) Wt. (g Moisture
(tare+solid) ) (tare+solid) (solid)
Al 0.9868 2.4139 1.4271 2.2346 1.2478 12.56
A2 1.0188 2.5639 1.5451 2.3583 1.3395
13.31
B1 1.0224 2.4077 1.3853 2.1975 1.1751
15.17
B2 1.0190 2.6346 , 1.6156 2.3693 1.3503
16.42
Cl _ 1.0094 1.7719 0.7625 1.5222 0.5128
32.75
C2 1.0206 1.6979 0.6773 1.4789 0.4583
32.33
(Samples were weighed, dried overnight @ 105 C without vacuum, cooled in
a desiccator, and then reweighed.)
Example 11 (K2SO4(s) Process Control Methods)
This example determined the range of process conditions in which
K2504(s) could be continuously produced without forming an adherent scale in
the process and the procedures to use to control sulfate solid formation and
io avoid a temporary shut down.
Precipitation of potassium sulfate in the sulfate circuit is a concern
because the process is based on liquid-liquid extraction and solids formation
could plug piping causing a maintenance action or even temporary shut down
for water washing recovery of the stripper (most likely S1-504 stage and E-
15 phase over-flow lines from the last metal sulfate strip unit (normally
S2M or
S3M or wash unit), to 51-504, then S1-504 to S2-504). During initial
experiments, the K2504(s) hard crystals plugged up the system and made the
equipment inoperable. The operation had to be temporarily shut down and
partially disassembled to remove the solid build-up.
zo The potassium sulfate crystallization reaction is shown as follows:
21C+ (aq) + S042- (aq)4.--- K2 SO4 (S) (20)
The solubility of potassium sulfate at ambient room temperature is
approximately 10.7 Wo by weight or 0.672 M when stoichiometric amounts of
potassium and sulfate ions are present. The literature data indicates that
25 K2504 solubility is decreased more by having excess K+ ion than by
reducing
temperature. Hence, we took this route to devise a K2504 solid production
process.
119
CA 02911119 2015-11-03
WO 2008/100610 PCT/US2008/002092
The solubility product expression for the reaction was calculated from:
K K's . [I( +12 [S042-1 (21)
Using the literature solubility data, the solubility product constant, Ksp,
was
calculated to be 1.212 M. By rearranging this equation, the sulfate ion
concentration can be calculated using
KsKr,SO,
{so42- ={K (22)
This equation shows that the sulfate ion concentration is set by the
solubility of K2SO4(s) and the sulfation solubility is set by the potassium
ion
concentration which means that the potassium and sulfate ion concentrations
lo need to be monitored in the sulfate circuit, especially in S1-SO4 and S2-
SO4
aqueous phases to avoid exceeding the K2SO4 solubility in the process which
would lead to the operating problems described above. Therefore, the AMD
treatment unit was operated at approximately 6-8% potassium carbonate by
weight fed to S4-SO4 to prevent crystallization of solids to avoid potential
problems such as plugging of the lines within the system, although 9% is still
effective.
By keeping the potassium ion concentration constant across the sulfate
strippers, the sulfate max was controlled at 80-90% of 1.212(e)2. The
potassium ion concentration is set by the K2SO4 harvest filtrate which was set
to 5.5% K2CO3 (0.500 M) and 6.5% K2SO4 (0.392 M). The maximum sulfate
concentration tolerable can be calculated based upon the filtrate potassium
ion concentration using Equation 22. This sulfate ion maximum concentration
limits the amount of sulfate ion that can be acquired in the sulfate strippers
and therefore can be used to prevent unwanted premature crystallization.
It is possible to control the potassium ion concentration by controlling the
sulfate and carbonate ion concentrations as seen by the charge balance in
=
Equation 23.
[1-1= [=-] (23)
[K ]= 2[S042-1+ *032-1+ [HCO3
120
CA 02911119 2015-11-03
27728-2D1
The carbonate and bicarbonate concentrations are related by pKa and can be
determined by atkatinity titration to pH 8.3 and then to pH 4.5., respectively
using standard HCR solution. By deriving the bicarbonate and carbonate
concentration equations, the potassium ion concentration can be calculated as
s shown by Equation 24.
C .1= 21SC )42- 14-12(V a )] cH I"- XN a) ¨ XIV c 11 (24)
By inserting Equation 24 into Equation 22, the process control Equation 25
was developed where KsKp2s04 =1.212M3.
K.sKpiso4
[S0:1, = ___________________________________________ (25)
ar [(2 [S042- 1) + v r;cII 3 xr
LrAcci )+
Therefore a control method includes one or more of the below:
1. Minimize the r exposure to the process using the process control
Equation 25.
2. Purge S1-504 aqueous phase at a rate to maintain (S042)sample at 80-
90% of (S042)max.
3. Be sure that none of the aqueous streams in the sulfate circuit exceeds
the comfort safety factor of 80-90% of (S042)max
4. Add K2CO3(aq) to S4- SO4 such that S3- SO4 and S4- 504 aqueous
phases are 570% of (S042)max and 5.1% of (S042)max respectively.
5. Because the operating window is narrow (about 10% percent), each
batch of filtrate should be assayed prior to introduction to the
operation at the S2- SO4 point.
While the forms of the invention herein disclosed constitute presently
preferred embodiments, many others are possible. It is not intended herein
to mention all of the possible equivalent forms or ramifications of the
invention. It is to be understood that the terms used herein are merely
descriptive, rather than limiting, and that various changes may be made
without departing from the scope of the claims.
121